Här beskrivs hur man upprättar och utför en kolorimetrisk analys för att bestämma koncentrationen av ett ämne i lösning.
Allmänt tillvägagångssätt
Vi kan inte placera ett material under ett mikroskop och räkna antalet molekyler per volymenhet på samma sätt som vi kan räkna antalet celler per volymenhet. Vi måste hitta något som vi kan mäta och som är proportionellt mot koncentrationen av det ämne som är intressant. Det mått som oftast används i analyser är absorbans av ljus. Beers lag säger oss att om en lösta substans absorberar ljus med en viss våglängd är absorbansen direkt proportionell mot koncentrationen av substansen i lösningen. En anordning som kallas spektrofotometer används för att mäta och visa och/eller registrera absorbansen i kvantifierbara enheter. Ofta absorberar inte ämnet i sig självt ljus för att möjliggöra en praktisk analys. Vi kan behöva använda ett eller flera reagenser för att producera färgade föreningar i proportion till koncentrationen av den okända substansen.
Mätning av ljusabsorptionen hos ett prov säger oss mycket lite om vi inte har en standard för jämförelse. Om till exempel prov X visar en absorbans på 0,5, vad är då den faktiska koncentrationen av X? Om vi har ett prov med känd koncentration och det provet också ger en absorbans på 0,5 är vi ganska säkra på att ämnet har samma koncentration. Anta att du har ett antal prover och att deras koncentrationer varierar. Det skulle vara användbart att ha ett antal standarder som täcker hela det sannolika koncentrationsområdet för vår okända substans. Det är där en standardkurva kommer in i bilden. Vi förbereder en serie standarder med känd koncentration av X, som sträcker sig från låg till hög koncentration. Vi kör analysen och plottar absorbansen mot koncentrationen för varje standard. Med hjälp av denna standardkurva kan vi avläsa koncentrationen för en okänd utifrån dess absorbansavläsning.
Kontroller
När vi utför en analys måste vi se till att endast den substans vi analyserar står för absorbansen av ljuset i det intressanta våglängdsområdet. Alla förhållanden under vilka standarder och okända ämnen framställs bör hållas identiska. Om lösningsmedel i provbuffertarna påverkar absorbansen har vi ett problem. Vi kommer inte att få korrekta resultat om vi varierar de volymer i vilka vi förbereder och analyserar standarder och okända ämnen. Tidpunkten för avläsning av absorbansen, temperaturen vid vilken vi förvarar materialen och alla andra fysiska faktorer bör hållas lika. Eftersom det inte alltid är praktiskt möjligt att använda identiska buffertar för alla okända ämnen och standarder behöver vi bara se till att ingen av komponenterna i någon av buffertarna har en betydande effekt på absorbansen.
När vi använder samma volym för alla standarder och okända ämnen förenklar vi analysen avsevärt. Standardkurvan kan plotta absorbansen mot ämnesmängden i stället för mot koncentrationen. Det kan vara mindre förvirrande att arbeta med mängder när man gör en analys, särskilt om det krävs utspädningar. Så länge du känner till den ursprungliga provvolymen som användes i en analys är det lätt att bestämma koncentrationen.
Komplikation
Alla analyser har gränser. Mängder av substans som understiger ett visst minimum kommer att vara odetekterbara. Över en viss maximal mängd eller koncentration blir en analys mättad, det vill säga ökningar av mängden eller koncentrationen påverkar inte absorbansen. Vi försöker i allmänhet arbeta inom det linjära området för en analys, dvs. där absorbansen är direkt proportionell mot koncentrationen. Helst skulle vi ställa in standarder som omfattar hela det användbara området för en analys. Det vill säga, vi optimerar analysens intervall.
Ofta är ett prov så koncentrerat att när man analyserar den föreskrivna provvolymen är resultatet utanför skalan – analysreagenset är mättat. Lösningen är då att späda ut provet. Om t.ex. volymen av varje standard eller prov är 1 ml, och 1 ml av din okända ger ett resultat som är utanför skalan, kan du tillsätta 0,1 ml prov till ett provrör tillsammans med 0,9 ml buffert. Om du läser av en koncentration från standardkurvan multiplicerar du resultatet med 10 för att få fram den faktiska koncentrationen i provet. Om du läser av en mängd från standardkurvan dividerar du helt enkelt den mängden med 0,1 ml för att få din koncentration.
När proverna är så koncentrerade att du inte kan pipettera en tillräckligt liten mängd med noggrannhet kan du bli tvungen att utföra serieutspädningar.
Exempel: framställning av en standardkurva
Vi kommer att sätta upp en hypotetisk analys för att mäta ämnet X. När X blandas med analysreagens bildas ett komplex som absorberar ljus vid våglängden 400 nm. Vår spektrofotometer kräver att vi lägger 2 ml volym i varje kuvett. En kuvett är ett genomskinligt kärl som placeras i en ljusbana för mätning av absorbansen. För att få rätt proportion mellan analysreagens och prov gör vi vår provvolym till 0,5 ml och tillsätter 1,5 ml färgreagens i varje rör. Om testet sätts upp på detta sätt kan man upptäcka mängder av X på så lite som 10 mikrogram (µg) till så mycket som 2 milligram (mg).
Referens
För att kalibrera spektrofotometern behöver vi ett referensrör som är identiskt i alla avseenden med standarderna och proverna, förutom att det inte innehåller någon substans X. Med ljusbanan blockerad kommer spektrofotometern att ställas in så att den avläser oändlig absorbans (ingen överföring av ljus överhuvudtaget). Med referensröret i ljusvägen ställer vi in spektrofotometern så att den avläser noll absorbans. På så sätt kommer ett prov som innehåller X att ge en absorbans inom det området. Referensröret används för att ge oss det maximala dynamiska området.
För detta hypotetiska exempel kommer referensen att innehålla 0,5 ml provbuffert och 1,5 ml färgreagens.
Standarder
Detta exempel beskriver en hypotetisk analys endast i illustrationssyfte.
Vi vill ha den bästa noggrannhet vi kan få, och vårt intervall sträcker sig över två storleksordningar, så ett sätt att ställa in standardkurvan är med en logaritmisk progression av standarder. Vi behöver standarder från 0,01 mg till 2 mg. Låt oss prova mängder på 0,01, 0,02, 0,05, 0,1, 0,2, 0,5, 1 och 2 mg. Den sista luckan är ganska stor, så vi tar med en standard på, låt oss säga, 1,5 mg. För att förbereda standarder är det lämpligt att börja med en koncentrerad stamlösning av ämnet. Den största mängd som vi behöver är 2 mg, i en volym på 0,5 ml. För att ge oss lite ”svängrum” kan vi göra en stamlösning på 5 mg/ml substans X. Följande tabell visar beräkningarna.
Tabell 1. Exempel på hur man planerar en standardkurva. Koncentrationen av protein i stamlösningen var 5 mg/ml. Detta exempel är endast avsett för illustrationsändamål.
| mängd substans X (mg) | volym stamlösning (µl) | volym buffert (µl) |
|
0 (referens) |
0 | 500 |
| 0.01 | 2 | 498* |
| 0.02 | 4 | 496* |
| 0.05 | 490 | |
| 0.1 | 20 | 480 |
| 0.2 | 40 | 460 |
| 0.5 | 100 | 400 |
| 1 | 200 | 300 |
| 1.5 | 300 | 200 |
| 2 | 400 | 100 |
*Det är vanligt att använda pipettrar som ger oss volymer som är exakta med högst två signifikanta siffror.
Volymen buffert är inte lika kritisk som volymen stamlösning. Fel i pipetteringen av buffert påverkar den totala volymen och därmed koncentrationen av färgreagenset. Fel på mindre än 1 % kommer inte att ha någon betydande inverkan på resultaten. Faktum är att om volymen av färgreagenset vida överstiger volymen av provet (inte i det här fallet) behöver vi inte ens utjämna volymerna genom att tillsätta buffert.
Vissa laboratorier är inte utrustade med pipettrar som går under 5 µl med noggrannhet. Det kan vara nödvändigt att utföra en serieutspädning för att få in till exempel 2 eller 4 µl stamlösning i ett analysrör.
Provberedning
Det underlättar att ha en rimlig uppskattning av de koncentrationsintervall för provet som man kan förvänta sig. Även med en sådan uppskattning är det bra att förbereda prover med en rad utspädningar, ifall ett prov är så koncentrerat att dess absorbansavläsningar ligger utanför intervallet.
För analysen i exemplet, om vi använder 500 µl prov i ett analysrör (den maximala volymen), måste dess koncentration vara mindre än 4 mg/ml för att ge en läsbar absorbans. Å andra sidan skulle vi vilja ha lika mycket om provet var t.ex. tio gånger mindre koncentrerat. Eftersom vi inte vet något om koncentrationen i ett visst prov skulle vi fylla ett rör med 500 µl för att täcka detta intervall. Eftersom analysen omfattar ett brett koncentrationsintervall kan vi använda 50 µl i ett andra analysrör. Nu kan provet vara så koncentrerat som 40 mg/ml och vi kommer fortfarande att ha 4 mg eller mindre i analysröret, vilket ger ett läsbart resultat. För att täcka alla baser kan vi analysera ett tredje rör med endast 5 µl prov.
Kör analysen
När alla standard- och okända ämnen är klara har vi:
- 1 referensrör
- några standarder som täcker hela analysområdet
- två eller tre analysrör per prov som representerar en serie utspädningar
Det är dags att genomföra proceduren för färgutveckling, vilket kan vara så enkelt som att tillsätta ett färgreagens och låta proverna sitta i några minuter. När det är praktiskt möjligt bör behandlingen av varje standard och prov tidsbestämmas så att absorbansen avläses efter samma tidsintervall för varje rör. Instrumentet skall kalibreras och sedan skall absorbanserna tas för varje rör i tur och ordning. En standardkurva erhålls genom att plotta absorbansen mot mängden substans X. Om förhållandet är klart linjärt är en standardkurva inte ens nödvändig. Mängderna kan bestämmas med hjälp av interpolation. En kurva bör konstrueras första gången en analys används, för att kontrollera noggrannhet och linjäritet.
Exempel på en standardkurva
Här är hur plotten skulle kunna se ut i en labbanteckningsbok (eleven har uppenbarligen en utmärkt handstil). Förhållandet är inte perfekt linjärt, utan visar snarare ett typiskt utdöendemönster.
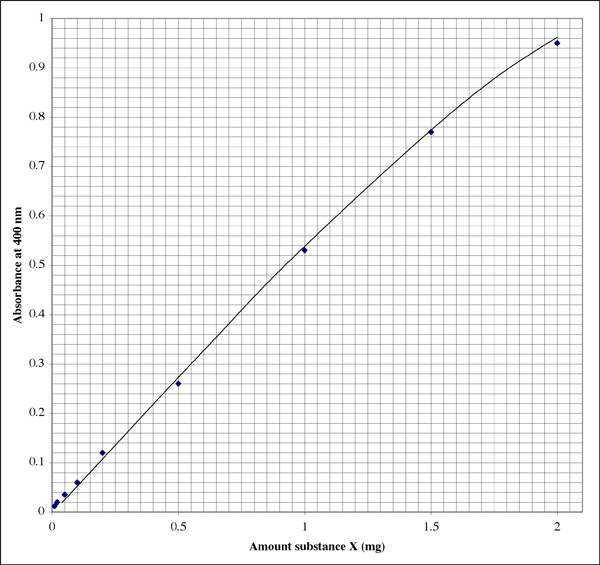
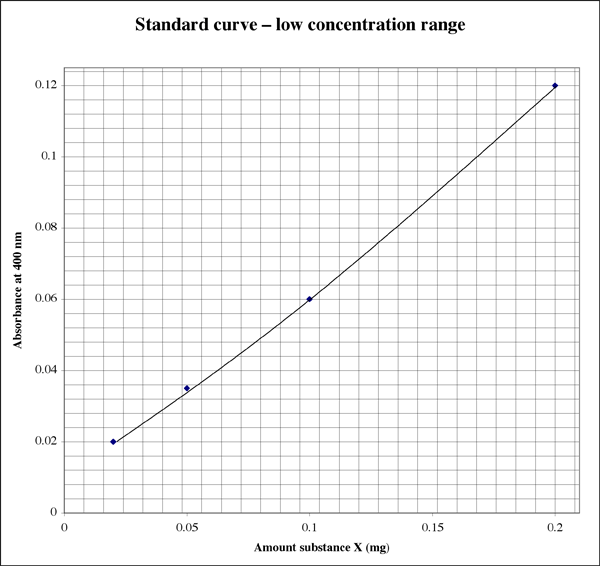
Då intervallet är så brett, för prover som ger mycket låga absorbansavläsningar kan en elev kanske vilja ha en andra plot med högre upplösning.
Bestäm koncentrationen av ett prov
En koncentration är en mängd av något per volymenhet. Vi rapporterar vanligtvis proteinkoncentrationer i milligram per milliliter (mg/ml), även om det ibland är bekvämt att använda mikrogram/mikroliter (µg/µl) eller kanske till och med µg/ml (för mycket små koncentrationer). För en okänd substans dividerar vi mängden substans (från standardkurvan) med volymen av det prov som används i analysen. Observera att denna volym inte är analysvolymen och inte heller den utspädda provvolymen. Dividera med volymen av det outspädda provet som du placerade i analysröret.
Antag att du har förberett tre analysrör för prov nr 1 som innehåller 500 µl, 50 µl respektive 5 µl prov. Anta att de gav absorbansvärden på 0,86, 0,12 respektive 0,01. Den sista absorbansen är förstås inte i skala. Interceptet bör vara noll, men vi kan inte räkna med att mycket låga absorbanser ger oss tillräckligt exakta avläsningar.\
En absorbans på 0,86 motsvarar 1,7 mg substans X. Volymen var 500 µl (0,5 ml), så vi får en koncentration på 3,4 mg/ml. Det låter bra. Vid kontroll av det andra läsbara röret visar en absorbans på 0,12 att röret innehöll 0,20 mg substans X. Volymen var 50 µl (0,050 ml). Koncentrationen bör vara 0,20 mg/0,050 ml = 4,0 mg/ml. Vilket resultat använder vi, eller tar vi ett genomsnitt?
Jag har funnit att man får de mest exakta resultaten genom att använda den absorbansavläsning som ligger närmast mitten av det känsliga området. I exemplet ovan är mitten en asorbans på 0,5, vilket motsvarar 0,1 mg substans. Absorbansskalan är logaritmisk, så även från en digital display är avläsningarna mer tillförlitliga i den nedre delen av skalan. Vid mycket låga absorbanser kommer emellertid en eller flera okända faktorer, t.ex. en defekt i provröret eller kyvetten, att ha en mer djupgående effekt på absorbansvärdet än vid högre absorbanser. I den övre delen av skalan närmar sig färgreagenset mättnad, vilket innebär att man inte bara får en sämre upplösning mellan absorbansavläsningarna, utan att reagenset också är mindre känsligt för skillnader i proteinkoncentrationen.