Definicja ko-delecji 1p/19q
Kompletna delecja zarówno krótkiego ramienia chromosomu 1 (1p), jak i długiego ramienia chromosomu 19 (19q) (ko-delecja 1p/19q) jest molekularną sygnaturą genetyczną oligodendroglejaków, podtypu pierwotnych guzów mózgu stanowiących około dziesięciu do piętnastu procent wszystkich glejaków rozsianych u dorosłych 1,2. Utrata jednego chromosomu hybrydowego powoduje utratę heterozygotyczności 1p i 19q (LOH) 3. Ta zmiana molekularna jest wynikiem niezrównoważonej translokacji całoramiennej między chromosomami 1 i 19 3 z utratą pochodnej t(1p;19q), która występuje we wczesnym okresie patogenezy oligodendroglejaków. Opisana po raz pierwszy w 1994 roku 4, biologiczny efekt ko-delecji 1p/19q pozostaje niejasny. Ko-delecja 1p/19q jest cennym biomarkerem diagnostycznym, prognostycznym i predykcyjnym w postępowaniu w nowotworach oligodendroglejowych.
Kolejna delecja 1p/19q jako biomarker diagnostyczny w glejaku
Kolejna delecja 1p/19q jest biomarkerem patognomonicznym, który definiuje odrębną jednostkę glejaka 5 i jest charakterystyczna dla oligodendroglejaków 6,7. Praktycznie wszystkie oligodendroglejaki z ko-delecją 1p/19q mają mutację w dehydrogenazie izocytanowej 1 (IDH1) przy argininie 132 (R132) lub analogicznej reszcie argininowej 172 w IDH2 (R172) 7,8. Inne częste zmiany molekularne współwystępujące z ko-delecją 1p/19q obejmują mutacje w promotorze genu odwrotnej transkryptazy telomerazy (TERT), mutacje w homologii Drosophila capicua (CIC) i far upstream element binding protein (FUBP1) 9 oraz metylację promotora genu metylotransferazy metylo-guaninowej (MGMT) 8,7. Z nielicznymi wyjątkami ko-delecja 1p/19q wyklucza się wzajemnie z mutacjami TP53 i ATRX, które charakteryzują guzy glejowe o linii astrocytarnej.
W związku z tym ocena ko-delecji 1p/19q, wraz ze statusem mutacji IDH i innymi markerami molekularnymi (np. ATRX i TP53), może pomóc w odróżnieniu oligodendroglejaków, które są zmutowane IDH i z delecją 1p/19q, od guzów z linii astrocytarnej, które nie mają ko-delecji 1p/19q 6,5.
Z klinicznego punktu widzenia, oligodendroglejaki z delecją 1p/19q są przeważnie guzami wieku dorosłego, ze szczytem zachorowań między czwartą a szóstą dekadą życia. Mają tendencję do występowania z napadami drgawek i zwykle obejmują płat czołowy, a niektóre guzy zawierają zwapnienia, które są wykrywalne w badaniach obrazowych mózgu.
Kolejna delecja 1p/19q jako biomarker prognostyczny w glejaku
Występowanie ko-delecji 1p/19q jest silnym niezależnym biomarkerem prognostycznym związanym z poprawą przeżycia zarówno w rozproszonych nowotworach o niskim stopniu złośliwości, jak i w nowotworach anaplastycznych 3,7,10,11,12.
Wśród wszystkich rozproszonych glejaków pacjenci z ko-delecją 1p/19q mają najkorzystniejsze rokowanie 13,7,14. W dużej multiomicznej i klinicznej analizie retrospektywnej przeprowadzonej przez The Cancer Genome Atlas Research Network (TCGA), pacjenci z glejakami stopnia II/III z mutacją IDH i ko-delecją 1p/19q mieli medianę przeżycia całkowitego wynoszącą 8,0 lat, w porównaniu z 6.3 lata u pacjentów z mutacją IDH i brakiem ko-delecji 1p/19q oraz 1,7 roku u pacjentów z dzikim typem IDH 7.
Kolejna delecja 1p/19q jako biomarker predykcyjny w glejaku
Kolejna delecja 1p/19q ma wartość predykcyjną dla odpowiedzi na chemioterapię w anaplastycznych oligodendroglejakach. Przedłużona obserwacja dwóch dużych randomizowanych badań kontrolowanych, w których porównywano chemioterapię prokarbazyną/lomustyną/winkrystyną (PCV) w połączeniu z radioterapią z samą radioterapią, wykazała przewagę przeżycia po zastosowaniu chemioterapii pierwszej linii w oligodendroglejakach z delecją 1p/19q 15,16. Długoterminowa analiza badania EORTC 26951, w którym 368 chorych poddano randomizacji do radioterapii lub radioterapii z PCV, wykazała, że mediana przeżycia całkowitego chorych z guzami z delecją 1p/19q wynosiła 9,3 roku w grupie leczonej samą radioterapią, ale nie została osiągnięta w grupie otrzymującej radioterapię z PCV 15. W podobnej analizie badania RTOG 9402, w którym 289 pacjentów otrzymało radioterapię lub PCV, a następnie radioterapię, mediana przeżycia całkowitego podwoiła się z 7,3 do 14,7 lat u pacjentów z guzami z delecją 1p/19q, którzy otrzymali PCV i radioterapię 16.
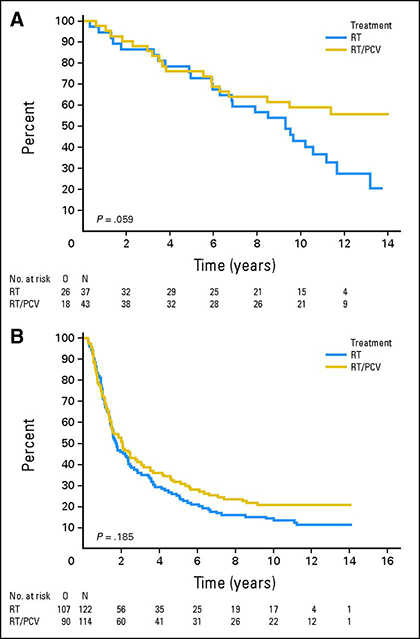
Overall survival for patients treated with radiotherapy (RT, blue curves) or radiotherapy plus PCV (RT/PCV, yellow curves) in patients with 1p/19q co-deleted (A) or non-co-deleted (B) tumours 15.
Podobnie, pacjenci z oligodendroglioma II stopnia „wysokiego ryzyka” (tj. pacjenci w wieku poniżej 40 lat, którzy przeszli resekcję subtotalną lub biopsję lub pacjenci w wieku 40 lat lub starsi), którzy otrzymali radioterapię plus PCV mieli dłuższe przeżycie całkowite w porównaniu z pacjentami leczonymi samą radioterapią w randomizowanym badaniu III fazy RTOG 9802 17.
Badanie ko-delecji 1p/19q w glejaku
Według klasyfikacji guzów ośrodkowego układu nerwowego Światowej Organizacji Zdrowia (WHO) z 2016 roku, ostateczne rozpoznanie oligodendroglejaka stopnia II i stopnia III (anaplastycznego) wymaga wykazania zarówno mutacji w rodzinie genu IDH, jak i ko-delecji 1p/19q 6. Istnieje kilka różnych metod stosowanych do identyfikacji pacjentów, których guzy są nosicielami delecji 1p/19q-co i nadal nie ma jasnego konsensusu co do tego, jak należy podchodzić do badań.
Hybrydyzacja fluorescencyjna in situ (FISH) jest wiarygodną, opłacalną metodą, która jest w stanie wykryć nieprawidłowość w minimalnych ilościach komórek nowotworowych na utrwalonych w formalinie, zatopionych w parafinie (FFPE) wycinkach tkanek. Podwójne sondy DNA znakowane fluorescencyjnie są używane do wykrywania loci 1p i 19q w jądrach interfazowych pojedynczych komórek glejaka z wycinków tkankowych FFPE przepisanych na niebarwione szkiełka. Zmiany w sygnałach sond 1p i 19q w porównaniu z kontrolami są wykorzystywane do określenia obecności delecji 1p/19q-co. Status chromosomu 1 i chromosomu 19 jest oceniany, na oddzielnych szkiełkach, poprzez analizę dystrybucji sond testowych i kontrolnych w 50-100 nienakładających się jądrach interfazowych 18. Wyniki FISH mogą być wyrażone jako odsetek komórek nowotworowych z usuniętym sygnałem lub jako stosunek sond testowych do kontrolnych dla każdego chromosomu 18. Wstępnie określone punkty odcięcia są wykorzystywane do określenia, czy delecja chromosomu jest obecna czy nie, a jeśli zarówno 1p jak i 19q są usunięte, zgłaszana jest ko-delecja 1p/19q.
Zmienność istnieje między obecnie dostępnymi sondami 1p i 19q FISH i loci chromosomowych, na które są ukierunkowane. Niektóre laboratoria stosują komercyjnie dostępne sondy FISH, podczas gdy inne ośrodki wytwarzają własne sondy we własnym zakresie18. Ponadto istnieją różnice w definicjach i punktach odcięcia stosowanych do określania delecji chromosomowej 18. Wyniki graniczne, które leżą blisko zdefiniowanego wcześniej punktu odcięcia dla delecji chromosomowej, mogą wymagać ponownego badania lub wyjaśnienia za pomocą dodatkowej techniki. Biorąc pod uwagę kliniczne znaczenie statusu delecji 1p/19q, ważne jest, aby protokoły FISH były standaryzowane w celu zapewnienia powtarzalnej oceny w ramach i pomiędzy laboratoriami. Komitet Badawczy Europejskiej Konfederacji Towarzystw Neuropatologicznych (Euro-CNS) opublikował praktyczne zalecenia wspomagające analizę opartą na FISH statusu 1p/19q 19.
FISH nie jest w stanie odróżnić delecji całego ramienia chromosomu z centromerowymi punktami przerwania charakterystycznymi dla oligodendroglejaków z delecją 1p/19q od mniejszych delecji ogniskowych. To rozróżnienie jest ważne, biorąc pod uwagę związek ko-delecji całego ramienia 1p/19q z poprawą przeżycia i odpowiedzią na chemioterapię w podtypie guza oligodendroglejowego. Array comparative genomic hybridisation (aCGH) i single nucleotide polymorphism (SNP) array są w stanie zidentyfikować utratę całego ramienia 1p lub 19q z większą wiarygodnością i mogą być stosowane zamiast FISH, jeśli jest to wykonalne. Jednakże, w porównaniu z FISH, techniki te są bardziej pracochłonne i wymagają wyższego odsetka komórek nowotworowych 18.
aCGH jest techniką wykrywania zmian liczby kopii genomu na poziomie wysokiej rozdzielczości. DNA – wyekstrahowane z próbki badanej i kontrolnej – są znakowane przy użyciu różnych barwników fluorescencyjnych, mieszane razem i hybrydyzowane do kilku tysięcy sond. Systemy obrazowania cyfrowego są używane do ilościowej oceny intensywności fluorescencji znakowanych sond DNA, które zhybrydyzowały do każdej sondy. Stosunek fluorescencji sygnałów hybrydyzacji testowej i kontrolnej jest określany w różnych pozycjach wzdłuż genomu, dostarczając informacji o względnej liczbie kopii sekwencji w badanym genomie w porównaniu do normalnego genomu. Metoda ta umożliwia jednoczesne wykrywanie aneuploidii chromosomowych, delecji, duplikacji i/lub amplifikacji dowolnego locus naniesionego na matrycę.
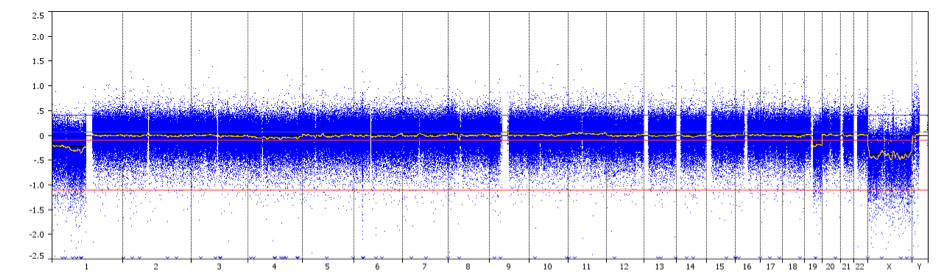
profil CGH przedstawiający całoramienną ko-delecję 1p/19q z centromerowymi punktami przerwania (strzałka) u pacjenta z oligodendroglioma stopnia II.
Kredyt: Prof. Marc Sanson.
Wreszcie, analiza mikrosatelitarna oparta na łańcuchowej reakcji polimerazy – umożliwiająca wykrycie LOH w wybranych loci, oraz sekwencjonowanie nowej generacji – metody oparte na NGS również mogą być stosowane do oceny statusu 1p/19q.
Wybór pacjentów
Wytyczne europejskie zalecają ocenę ko-delecji1p/19q w celu potwierdzenia rozpoznania oligodendroglioma oraz przewidywania chemiowrażliwości i rokowania u tych chorych 5,20. Na podstawie wyników badań EORTC 26951 i RTOG 9402 pacjenci z anaplastycznymi guzami oligodendroglejowymi z delecją 1p/19q nie powinni być leczeni wyłącznie radioterapią, ale zamiast tego powinni otrzymać wczesną chemioterapię alkilującą z radioterapią 20. Podobne podejście należy rozważyć w podgrupie pacjentów z oligodendroglioma w stopniu II.
W przypadku konieczności rozpoczęcia leczenia farmakologicznego wielkość korzyści z leczenia skojarzonego radioterapia plus PCV jest znaczna, ale działania toksyczne są większe u pacjentów otrzymujących takie połączenie. Trwają badania kliniczne oceniające wartość samej chemioterapii upfront lub radioterapii skojarzonej plus temozolomid w porównaniu ze standardową radioterapią plus PCV.
Key References
- Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a „state of the science” review. Neuro Oncol 2014; 16(7):896-913.
- Ibdaih A, Marie Y, Pierron G, et al. Two types of chromosome 1p losses with opposite significance in gliomas. Ann Neurol 2005; 58(3):483-7.
- Jenkins R, Blair H, Ballman K, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 2006; 66: 9852-61.
- Reifenberger J, Reifenberger G, Liu L, et al. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 1994; 145:1175-90.
- Stupp R, Brada M, van den Bent M, et al. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 (Suppl 3): iii93-101.
- Louis D, Perry A, Reifenberger G. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathologica 2016; 131: 803-20.
- Cancer Genome Atlas Research Network, Brat D, Verhaak R, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 2015; 372: 2481-98.
- Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009; 27: 4150-54.
- Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011; 333(6048): 1453-5.
- Zhao J, Ma W, Zhao H. Loss of heterozygosity 1p/19q and survival in glioma: a meta-analysis. Neuro-Oncology 2014; 16: 103-112.
- van den Bent M, Carpentier A, Brandes A, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 2006; 24: 2715-22
- Cairncross G, Berkey B, Shaw E, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 2006; 24: 2707-14.
- Wiestler B, Capper D, Sill M, et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathologica 2014; 128:561-71.
- Ceccarelli M, Barthel FP, Malta TM, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016; 164(3): 550-63.
- van den Bent M, Brandes A, Taphoorn M, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 2013; 31:344-50.
- Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol 2013; 31: 337-43.
- Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N Engl J Med 2016; 374: 1344-1355.
- Woehrer A, Sander P, Baberler C, et al. FISH-based detection of 1p 19q codeletion in oligodendroglial tumors: procedures and protocols for neuropathological practice – a publication under the auspices of the Research Committee of the European Confederation of Neuropathological Societies (Euro-CNS). Clinical Neuropathology 2011; 30: 47-55.
- Pinkham M, Telford N, Whitfield G, et al. FISHing Tips: What Every Clinician Should Know About 1p19q Analysis in Gliomas Using Fluorescence in situ Hybridisation. Clinical Oncology 2015; 27: 445-53.
- Weller M, van den Bent M, Hopkins K, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol 2014; 15: 395-403.
.