Definition der 1p/19q-Kodeletion
Die vollständige Deletion sowohl des kurzen Arms von Chromosom 1 (1p) als auch des langen Arms von Chromosom 19 (19q) (1p/19q-Kodeletion) ist die molekulargenetische Signatur von Oligodendrogliomen, einer Unterart primärer Hirntumoren, die etwa zehn bis fünfzehn Prozent aller diffusen Gliome bei Erwachsenen ausmachen 1,2. Der Verlust eines Hybridchromosoms führt zu einem Verlust der Heterozygotie (LOH) von 1p und 19q 3. Diese molekulare Veränderung ist das Ergebnis einer unausgewogenen ganzarmigen Translokation zwischen den Chromosomen 1 und 19 3 mit dem Verlust des Derivats t(1p;19q), die schon früh in der Pathogenese von Oligodendrogliomen auftritt. Die biologische Wirkung der 1p/19q-Kodeletion, die erstmals 1994 beschrieben wurde 4, ist nach wie vor unklar. Die 1p/19q-Ko-Deletion ist ein wertvoller diagnostischer, prognostischer und prädiktiver Biomarker für die Behandlung von Oligodendroglia-Tumoren.
1p/19q-Ko-Deletion als diagnostischer Biomarker bei Gliomen
Die 1p/19q-Ko-Deletion ist ein pathognomonischer Biomarker, der eine bestimmte Gliom-Entität definiert 5 und für Oligodendrogliome charakteristisch ist 6,7. Praktisch alle Oligodendrogliome mit 1p/19q-Koletion weisen eine Mutation in der Isocitrat-Dehydrogenase 1 (IDH1) an Arginin 132 (R132) oder dem analogen Rest Arginin 172 in IDH2 (R172) auf 7,8. Andere häufige molekulare Veränderungen, die mit einer 1p/19q-Kodeletion einhergehen, sind Mutationen im Promotor des Telomerase-Reverse-Transkriptase-Gens (TERT), Mutationen im Homolog von Drosophila capicua (CIC) und im Far Upstream Element Binding Protein (FUBP1) 9 sowie eine Methylierung des Promotors des Methyl-Guanin-Methyl-Transferase-Gens (MGMT) 8,7. Von wenigen Ausnahmen abgesehen, schließt sich die 1p/19q-Ko-Deletion gegenseitig mit TP53- und ATRX-Mutationen aus, die beide Glia-Tumoren astrozytärer Abstammung charakterisieren.
Daher ist die Bewertung der 1p/19q-Ko-Deletion zusammen mit dem IDH-Mutationsstatus und anderen molekularen Markern (z. B. ATRX- und TP53-Status) dazu beitragen, Oligodendrogliome, die IDH-mutiert und 1p/19q-kodeletiert sind, von Tumoren astrozytärer Abstammung zu unterscheiden, die nicht 1p/19q-kodeletiert sind.6,5.
Aus klinischer Sicht sind 1p/19q-kodeletierte Oligodendrogliome vorwiegend Tumoren des Erwachsenenalters, mit einem Häufigkeitsgipfel zwischen dem vierten und dem sechsten Lebensjahrzehnt. Sie treten in der Regel mit Krampfanfällen auf und betreffen typischerweise den Frontallappen, wobei einige Tumoren Verkalkungen enthalten, die in der Bildgebung des Gehirns nachweisbar sind.
1p/19q-Ko-Deletion als prognostischer Biomarker bei Gliomen
Das Vorhandensein einer 1p/19q-Ko-Deletion ist ein starker unabhängiger prognostischer Biomarker, der sowohl bei diffusen niedriggradigen als auch bei anaplastischen Tumoren mit einem verbesserten Überleben assoziiert ist 3,7,10,11,12.
Unter allen diffusen Gliomen haben Patienten mit 1p/19q-Ko-Deletion die günstigste Prognose 13,7,14. In einer großen multimikroskopischen und klinischen retrospektiven Analyse, die vom The Cancer Genome Atlas Research Network (TCGA) durchgeführt wurde, hatten Patienten mit Gliomen des Grades II/III mit einer IDH-Mutation und einer 1p/19q-Co-Deletion ein medianes Gesamtüberleben von 8,0 Jahren, verglichen mit 6.3 Jahren bei Patienten mit einer IDH-Mutation und keiner 1p/19q-Ko-Deletion und 1,7 Jahren bei Patienten mit Wildtyp-IDH 7.
1p/19q-Ko-Deletion als prädiktiver Biomarker bei Gliomen
1p/19q-Ko-Deletion hat prädiktiven Wert für das Ansprechen auf Chemotherapie bei anaplastischen Oligodendrogliomen. Die erweiterte Nachbeobachtung zweier großer randomisierter kontrollierter Studien, in denen eine Chemotherapie mit Procarbazin/Lomustin/Vincristin (PCV) in Kombination mit einer Strahlentherapie mit einer alleinigen Strahlentherapie verglichen wurde, zeigte einen Überlebensvorteil der Erstlinien-Chemotherapie bei Oligodendrogliomen mit 1p/19q-co-Deletion 15,16. Eine Langzeitanalyse der EORTC 26951-Studie, in der 368 Patienten randomisiert einer Strahlentherapie oder einer Strahlentherapie mit anschließender PCV unterzogen wurden, zeigte, dass die mediane Gesamtüberlebenszeit für Patienten mit 1p/19q-co-verarmten Tumoren bei 9,3 Jahren lag, wenn sie nur mit Strahlentherapie behandelt wurden, während sie bei Patienten, die Strahlentherapie plus PCV erhielten, noch nicht erreicht wurde 15. In einer ähnlichen Analyse der RTOG 9402-Studie, in der 289 Patienten eine Strahlentherapie oder eine PCV gefolgt von einer Strahlentherapie erhielten, verdoppelte sich das mediane Gesamtüberleben von 7,3 auf 14,7 Jahre bei Patienten mit 1p/19q-co-deletierten Tumoren, die PCV und Strahlentherapie erhielten 16.
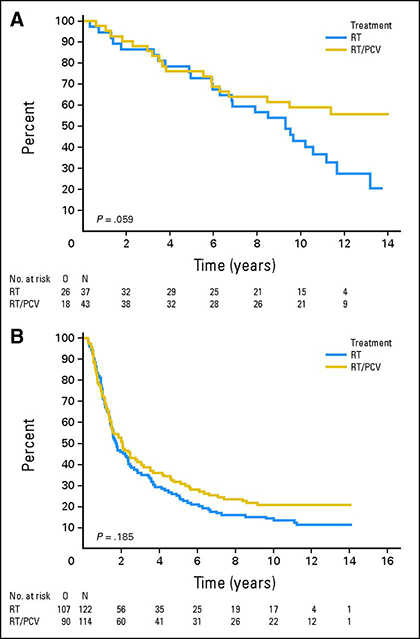
Gesamtüberleben für Patienten, die mit Strahlentherapie (RT, blaue Kurven) oder Strahlentherapie plus PCV (RT/PCV, gelbe Kurven) behandelt wurden, bei Patienten mit 1p/19q-ko-deletierten (A) oder nicht-ko-deletierten (B) Tumoren 15.
Auch in der randomisierten Phase-III-Studie RTOG 9802 hatten Patienten mit einem „Hochrisiko“-Oligodendrogliom vom Grad II (d. h. Patienten, die jünger als 40 Jahre alt waren und sich einer subtotalen Resektion oder Biopsie unterzogen hatten, oder Patienten, die 40 Jahre alt oder älter waren), die eine Strahlentherapie plus PCV erhielten, ein längeres Gesamtüberleben als Patienten, die nur mit Strahlentherapie behandelt wurden 17.
1p/19q-Ko-Deletionstest bei Gliomen
Nach der Klassifikation von Tumoren des Zentralnervensystems der Weltgesundheitsorganisation (WHO) von 2016 erfordert die endgültige Diagnose eines Oligodendroglioms vom Grad II und III (anaplastisch) den Nachweis sowohl einer Mutation in der IDH-Genfamilie als auch einer 1p/19q-Ko-Deletion 6. Es gibt verschiedene Methoden zur Identifizierung von Patienten, deren Tumoren eine 1p/19q-co-Deletion aufweisen, und es besteht immer noch kein eindeutiger Konsens darüber, wie die Tests durchgeführt werden sollten.
Die fluoreszierende In-situ-Hybridisierung (FISH) ist eine zuverlässige, kostengünstige Methode, mit der die Anomalie in minimalen Mengen von Tumorzellen auf formalinfixierten, paraffineingebetteten (FFPE) Gewebeschnitten nachgewiesen werden kann. Es werden zweifach fluoreszenzmarkierte DNA-Sonden verwendet, um die 1p- und 19q-Loci in den Interphasenkernen einzelner Gliomzellen aus FFPE-Gewebeschnitten nachzuweisen, die auf ungefärbte Objektträger übertragen wurden. Veränderungen in den 1p- und 19q-Sondensignalen im Vergleich zu den Kontrollen werden zur Bestimmung des Vorhandenseins einer 1p/19q-Co-Deletion verwendet. Der Status von Chromosom 1 und Chromosom 19 wird auf separaten Objektträgern durch Analyse der Verteilung von Test- und Kontrollsonden in 50-100 nicht überlappenden Interphasenkernen ermittelt 18. Die FISH-Ergebnisse können als Prozentsatz der Tumorzellen mit einem Deletionssignal oder als Verhältnis von Test- zu Kontrollsonden für jedes Chromosom 18 angegeben werden. Anhand vorgegebener Grenzwerte wird festgestellt, ob eine Chromosomen-Deletion vorliegt oder nicht, und wenn sowohl 1p als auch 19q deletiert sind, wird eine 1p/19q-Ko-Deletion gemeldet.
Die derzeit verfügbaren 1p- und 19q-FISH-Sonden und die Chromosomenloci, auf die sie abzielen, sind unterschiedlich. Einige Labors verwenden kommerziell erhältliche FISH-Sonden, während andere Zentren ihre eigenen Sonden selbst herstellen18. Darüber hinaus gibt es Unterschiede bei den Definitionen und Grenzwerten für die Bestimmung der Chromosomen-Deletion 18. Grenzwertige Ergebnisse, die nahe an einem vordefinierten Cut-off-Wert für eine Chromosomen-Deletion liegen, erfordern möglicherweise eine erneute Untersuchung oder eine Abklärung mit einer zusätzlichen Technik. In Anbetracht der klinischen Bedeutung des 1p/19q-Co-Deletionsstatus ist es wichtig, dass die FISH-Protokolle standardisiert werden, um eine reproduzierbare Bewertung innerhalb und zwischen den Labors zu gewährleisten. Das Forschungskomitee der European Confederation of Neuropathological Societies (Euro-CNS) hat praktische Empfehlungen zur Unterstützung der FISH-basierten Analyse des 1p/19q-Status veröffentlicht 19.
FISH ist nicht in der Lage, zwischen den Deletionen ganzer Chromosomenarme mit zentromerischen Bruchpunkten, die für Oligodendrogliome mit 1p/19q-Ko-Deletion charakteristisch sind, und kleineren fokalen Deletionen zu unterscheiden. Diese Unterscheidung ist wichtig, da die 1p/19q-Ko-Deletion mit einer verbesserten Überlebensrate und einem besseren Ansprechen auf die Chemotherapie beim Oligodendrogliom-Subtyp in Verbindung gebracht wird. Die vergleichende genomische Array-Hybridisierung (aCGH) und der Array für Einzelnukleotid-Polymorphismen (SNP) sind in der Lage, den Verlust des gesamten Arms von 1p oder 19q mit höherer Zuverlässigkeit zu identifizieren und können, wenn möglich, anstelle von FISH eingesetzt werden. Im Vergleich zu FISH sind diese Techniken jedoch arbeitsintensiver und erfordern einen höheren Anteil an neoplastischen Zellen.18
aCGH ist eine Technik zum Nachweis genomischer Kopienzahlvariationen mit hoher Auflösung. DNA – extrahiert aus einer Test- und einer Kontrollprobe – wird mit verschiedenen Fluoreszenzfarbstoffen markiert, zusammengemischt und mit mehreren tausend Sonden hybridisiert. Digitale Bildgebungssysteme werden zur Quantifizierung der Fluoreszenzintensitäten der markierten DNA-Sonden verwendet, die mit jeder Sonde hybridisiert haben. Das Fluoreszenzverhältnis der Test- und Kontrollhybridisierungssignale wird an verschiedenen Positionen entlang des Genoms bestimmt und gibt Aufschluss über die relative Kopienzahl der Sequenzen im Testgenom im Vergleich zu einem normalen Genom. Diese Methode ermöglicht den gleichzeitigen Nachweis von Chromosomenaneuploidien, Deletionen, Duplikationen und/oder Amplifikationen eines beliebigen Locus, der auf einem Array aufgetragen ist.
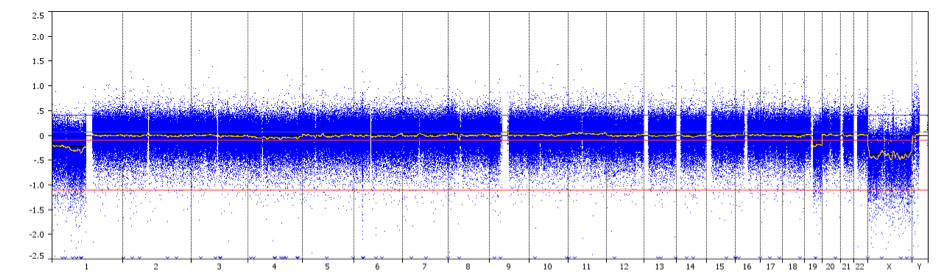
aCGH-Profil, das eine 1p/19q-Ganzarm-Ko-Deletion mit zentromerischen Bruchstellen (Pfeil) bei einem Patienten mit Oligodendrogliom Grad II zeigt.
Gutschrift: Prof. Marc Sanson.
Schließlich können die auf der Polymerase-Kettenreaktion basierende Mikrosatellitenanalyse – die den Nachweis von LOH an ausgewählten Loci ermöglicht – und NGS-basierte Methoden (Next Generation Sequencing) auch zur Bewertung des 1p/19q-Status verwendet werden.
Patientenauswahl
Europäische Leitlinien empfehlen, dass die 1p/19q-Kodeletion zur Unterstützung der Diagnose eines Oligodendroglioms und zur Vorhersage der Chemosensitivität und Prognose dieser Patienten bewertet werden sollte 5,20. Auf der Grundlage der Ergebnisse der Studien EORTC 26951 und RTOG 9402 sollten Patienten mit anaplastischen Oligodendroglia-Tumoren mit 1p/19q-co-Deletion nicht allein mit Strahlentherapie behandelt werden, sondern stattdessen eine frühe alkylierende Chemotherapie mit Strahlentherapie erhalten 20. Ein ähnlicher Ansatz sollte bei einer Untergruppe von Patienten mit Oligodendrogliom Grad II in Betracht gezogen werden.
Wenn eine medizinische Behandlung eingeleitet werden muss, ist das Ausmaß des Behandlungsnutzens einer kombinierten Strahlentherapie plus PCV beträchtlich, aber die toxischen Wirkungen sind bei Patienten, die diese Kombination erhalten, größer. Derzeit laufen klinische Studien, um den Wert einer alleinigen Upfront-Chemotherapie oder einer kombinierten Strahlentherapie plus Temozolomid im Vergleich zu einer Standard-Strahlentherapie plus PCV zu bewerten.
Key References
- Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a „state of the science“ review. Neuro Oncol 2014; 16(7):896-913.
- Ibdaih A, Marie Y, Pierron G, et al. Two types of chromosome 1p losses with opposite significance in gliomas. Ann Neurol 2005; 58(3):483-7.
- Jenkins R, Blair H, Ballman K, et al. Ein t(1;19)(q10;p10) vermittelt die kombinierten Deletionen von 1p und 19q und sagt eine bessere Prognose von Patienten mit Oligodendrogliom voraus. Cancer Res 2006; 66: 9852-61.
- Reifenberger J, Reifenberger G, Liu L, et al. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 1994; 145:1175-90.
- Stupp R, Brada M, van den Bent M, et al. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 (Suppl 3): iii93-101.
- Louis D, Perry A, Reifenberger G. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathologica 2016; 131: 803-20.
- Cancer Genome Atlas Research Network, Brat D, Verhaak R, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 2015; 372: 2481-98.
- Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009; 27: 4150-54.
- Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011; 333(6048): 1453-5.
- Zhao J, Ma W, Zhao H. Loss of heterozygosity 1p/19q and survival in glioma: a meta-analysis. Neuro-Oncology 2014; 16: 103-112.
- van den Bent M, Carpentier A, Brandes A, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 2006; 24: 2715-22
- Cairncross G, Berkey B, Shaw E, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 2006; 24: 2707-14.
- Wiestler B, Capper D, Sill M, et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathologica 2014; 128:561-71.
- Ceccarelli M, Barthel FP, Malta TM, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016; 164(3): 550-63.
- van den Bent M, Brandes A, Taphoorn M, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 2013; 31:344-50.
- Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol 2013; 31: 337-43.
- Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N Engl J Med 2016; 374: 1344-1355.
- Woehrer A, Sander P, Baberler C, et al. FISH-based detection of 1p 19q codeletion in oligodendroglial tumors: procedures and protocols for neuropathological practice – a publication under the auspices of the Research Committee of the European Confederation of Neuropathological Societies (Euro-CNS). Clinical Neuropathology 2011; 30: 47-55.
- Pinkham M, Telford N, Whitfield G, et al. FISHing Tips: What Every Clinician Should Know About 1p19q Analysis in Gliomas Using Fluorescence in situ Hybridisation. Clinical Oncology 2015; 27: 445-53.
- Weller M, van den Bent M, Hopkins K, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol 2014; 15: 395-403.