Définition de la codélétion 1p/19q
La délétion complète à la fois du bras court du chromosome 1 (1p) et du bras long du chromosome 19 (19q) (codélétion 1p/19q) est la signature génétique moléculaire des oligodendrogliomes, un sous-type de tumeurs cérébrales primaires représentant environ dix à quinze pour cent de tous les gliomes diffus chez les adultes 1,2. La perte d’un chromosome hybride entraîne une perte d’hétérozygotie (LOH) de 1p et 19q 3. Cette altération moléculaire est le résultat d’une translocation déséquilibrée à bras entier entre les chromosomes 1 et 19 3 avec la perte du dérivé t(1p;19q), qui se produit tôt dans la pathogenèse des oligodendrogliomes. Initialement décrite en 1994 4, l’effet biologique de la codélétion 1p/19q reste peu clair. La codélétion 1p/19q est un biomarqueur diagnostique, pronostique et prédictif précieux pour la gestion des tumeurs oligodendrogliales.
La codélétion 1p/19q comme biomarqueur diagnostique dans les gliomes
La codélétion 1p/19q est un biomarqueur pathognomonique qui définit une entité distincte de gliome 5 et est caractéristique des oligodendrogliomes 6,7. Pratiquement tous les oligodendrogliomes codélégués 1p/19q présentent une mutation de l’isocitrate déshydrogénase 1 (IDH1) au niveau de l’arginine 132 (R132) ou du résidu analogue, l’arginine 172, dans l’IDH2 (R172) 7,8. Parmi les autres altérations moléculaires courantes accompagnant la codélétion 1p/19q, citons les mutations du promoteur du gène de la télomérase transcriptase inverse (TERT), les mutations de l’homologue de la drosophile capicua (CIC) et de la protéine de liaison de l’élément situé très en amont (FUBP1) 9, et la méthylation du promoteur du gène de la méthyl-guanine méthyl-transférase (MGMT) 8,7. A quelques exceptions près, la codélétion 1p/19q est mutuellement exclusive avec la mutation de TP53 et d’ATRX, qui caractérisent toutes deux les tumeurs gliales de la lignée astrocytaire.
Par conséquent, l’évaluation de la codélétion 1p/19q, ainsi que le statut de la mutation de l’IDH et d’autres marqueurs moléculaires (par ex. ATRX et TP53), peut aider à distinguer les oligodendrogliomes mutants IDH et codéifiés 1p/19q, des tumeurs de la lignée astrocytaire non codéifiées 1p/19q 6,5.
D’un point de vue clinique, les oligodendrogliomes codéifiés 1p/19q sont principalement des tumeurs de l’âge adulte, avec un pic d’incidence entre la quatrième et la sixième décennie de vie. Ils ont tendance à se présenter avec des crises d’épilepsie et touchent généralement le lobe frontal, certaines tumeurs contenant des calcifications détectables à l’imagerie cérébrale.
La codélétion 1p/19q comme biomarqueur pronostique dans le gliome
La présence de la codélétion 1p/19q est un biomarqueur pronostique indépendant fort associé à une meilleure survie dans les tumeurs diffuses de bas grade et anaplasiques 3,7,10,11,12.
Parmi tous les gliomes diffus, les patients présentant une codélétion 1p/19q ont le pronostic le plus favorable 13,7,14. Dans une vaste analyse rétrospective clinique et multiomique réalisée par le réseau de recherche The Cancer Genome Atlas (TCGA), les patients atteints de gliomes de grade II/III présentant une mutation de l’IDH et une co-délétion 1p/19q avaient une survie globale médiane de 8,0 ans, contre 6.3 ans chez les patients présentant une mutation de l’IDH et aucune codélétion 1p/19q et 1,7 ans chez les patients présentant un IDH de type sauvage 7.
La codélétion 1p/19q comme biomarqueur prédictif dans le gliome
La codélétion 1p/19q a une valeur prédictive pour la réponse à la chimiothérapie dans les oligodendrogliomes anaplasiques. Le suivi prolongé de deux grands essais contrôlés randomisés qui ont comparé la chimiothérapie par procarbazine/lomustine/vincristine (PCV) en association avec la radiothérapie à la radiothérapie seule a démontré un avantage de survie de la chimiothérapie de première ligne dans les oligodendrogliomes à co-délétion 1p/19q 15,16. L’analyse à long terme de l’étude EORTC 26951, dans laquelle 368 patients ont été randomisés pour recevoir une radiothérapie ou une radiothérapie suivie d’une chimiothérapie par PCV, a montré que la survie globale médiane des patients atteints de tumeurs à co-détection 1p/19q était de 9,3 ans pour ceux traités par radiothérapie seule, mais qu’elle n’était pas encore atteinte pour ceux qui avaient reçu une radiothérapie plus une chimiothérapie par PCV 15. Dans une analyse similaire de l’essai RTOG 9402, dans lequel 289 patients ont reçu une radiothérapie ou un VCP suivi d’une radiothérapie, la survie globale médiane a doublé, passant de 7,3 à 14,7 ans, chez les patients atteints de tumeurs à co-délétion 1p/19q qui ont reçu un VCP et une radiothérapie 16.
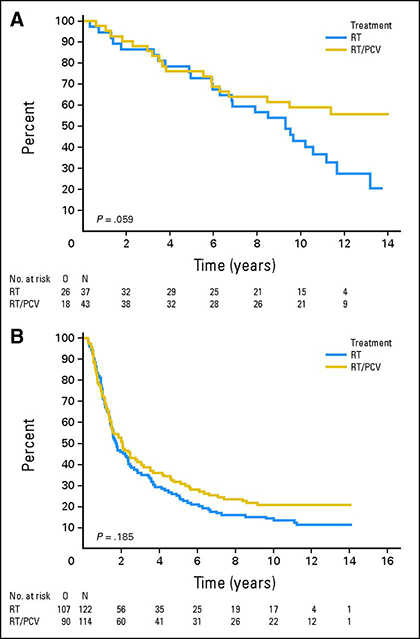
Survie globale des patients traités par radiothérapie (RT, courbes bleues) ou radiothérapie plus PCV (RT/PCV, courbes jaunes) chez les patients atteints de tumeurs 1p/19q co-délestées (A) ou non co-délestées (B) 15.
De même, les patients atteints d’un oligodendrogliome de grade II à « haut risque » (c’est-à-dire les patients âgés de moins de 40 ans ayant subi une résection subtotale ou une biopsie ou les patients âgés de 40 ans ou plus) qui ont reçu une radiothérapie plus PCV ont eu une survie globale plus longue que les patients traités par radiothérapie seule dans l’essai randomisé de phase III RTOG 9802 17.
Test de codélétion 1p/19q dans les gliomes
Selon la classification 2016 de l’Organisation mondiale de la santé (OMS) des tumeurs du système nerveux central, le diagnostic définitif d’oligodendrogliome de grade II et de grade III (anaplasique) nécessite la démonstration à la fois d’une mutation de la famille du gène IDH et d’une codélétion 1p/19q 6. Plusieurs méthodes différentes ont été utilisées pour identifier les patients dont les tumeurs présentent une codélétion 1p/19q et il n’y a toujours pas de consensus clair sur la façon dont le test doit être abordé.
L’hybridation in situ fluorescente (FISH) est une méthode fiable et rentable, capable de détecter l’anomalie dans des quantités minimes de cellules tumorales sur des coupes de tissus fixés au formol et inclus en paraffine (FFPE). Des sondes d’ADN à double marquage fluorescent sont utilisées pour détecter les loci 1p et 19q dans les noyaux en interphase de cellules de gliome individuelles provenant de coupes de tissus FFPE transcrites sur des lames non colorées. Les changements dans les signaux des sondes 1p et 19q par rapport aux contrôles sont utilisés pour déterminer la présence d’une co-délétion 1p/19q. Les statuts du chromosome 1 et du chromosome 19 sont évalués, sur des lames séparées, en analysant la distribution des sondes de test et de contrôle dans 50-100 noyaux interphasiques non chevauchants 18. Les résultats de la FISH peuvent être exprimés en pourcentage de cellules tumorales présentant un signal délété ou en rapport entre les sondes de test et de contrôle pour chaque chromosome 18. Des seuils de coupure pré-spécifiés sont utilisés pour déterminer si une délétion chromosomique est présente ou non, et si à la fois 1p et 19q sont délétés, une codélétion 1p/19q est signalée.
Il existe une variabilité entre les sondes FISH 1p et 19q actuellement disponibles et les loci chromosomiques qu’elles ciblent. Certains laboratoires utilisent des sondes FISH disponibles dans le commerce alors que d’autres centres fabriquent les leurs en interne 18. En outre, il existe des variations dans les définitions et les seuils utilisés pour déterminer la délétion chromosomique 18. Les résultats limites qui sont proches d’un seuil prédéfini pour la délétion chromosomique peuvent nécessiter un nouveau test ou une clarification avec une technique supplémentaire. Compte tenu de l’importance clinique du statut de co-délétion 1p/19q, il est important que les protocoles FISH soient standardisés afin de garantir une évaluation reproductible au sein des laboratoires et entre eux. Le comité de recherche de la Confédération européenne des sociétés de neuropathologie (Euro-CNS) a publié des recommandations pratiques pour faciliter l’analyse du statut 1p/19q par FISH 19.
La FISH est incapable de différencier les délétions de bras chromosomiques entiers avec des points de rupture centromériques caractéristiques des oligodendrogliomes codélégués 1p/19q, des délétions focales plus petites. Cette distinction est importante étant donné l’association de la codélétion du bras entier 1p/19q avec une meilleure survie et une meilleure réponse à la chimiothérapie dans le sous-type de tumeur oligodendrogliale. L’hybridation génomique comparative en réseau (aCGH) et les réseaux de polymorphisme nucléotidique unique (SNP) sont capables d’identifier la perte du bras entier de 1p ou 19q avec une plus grande fiabilité et peuvent être utilisés de préférence à la FISH lorsque cela est possible. Cependant, par rapport à la FISH, ces techniques ont tendance à demander plus de travail et nécessitent une proportion plus élevée de cellules néoplasiques 18.
aCGH est une technique pour détecter les variations du nombre de copies génomiques à un niveau de haute résolution. L’ADN – extrait d’un échantillon test et d’un échantillon témoin – est marqué à l’aide de différents colorants fluorescents, mélangé et hybridé à plusieurs milliers de sondes. Des systèmes d’imagerie numérique sont utilisés pour quantifier les intensités de fluorescence des sondes d’ADN marquées qui se sont hybridées à chaque sonde. Le rapport de fluorescence des signaux d’hybridation du test et du contrôle est déterminé à différentes positions le long du génome, fournissant des informations sur le nombre relatif de copies de séquences dans le génome testé par rapport à un génome normal. Cette méthode permet la détection simultanée d’aneuploïdies chromosomiques, de délétions, de duplications et/ou d’amplifications de n’importe quel locus tracé sur une matrice.
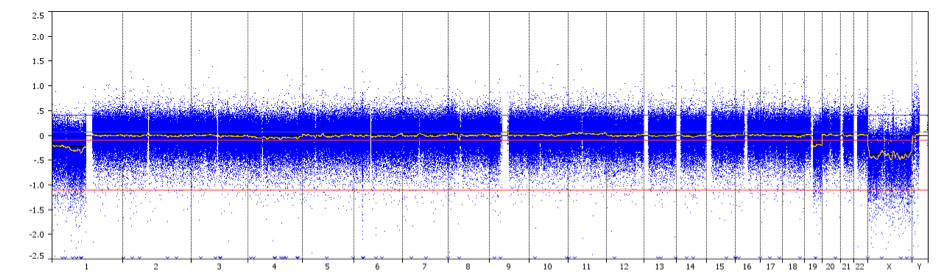
un profil CCGH montrant une codélétion 1p/19q sur tout le bras avec des points de rupture centromériques (flèche) chez un patient atteint d’un oligodendrogliome de grade II.
Crédit : Prof. Marc Sanson.
Enfin, l’analyse des microsatellites basée sur la réaction en chaîne par polymérase – qui permet de détecter la LOH à des loci sélectionnés, et les méthodes basées sur le séquençage de nouvelle génération – NGS – peuvent également être utilisées pour évaluer le statut 1p/19q.
Sélection des patients
Les directives européennes recommandent que la co-délétion 1p/19q soit évaluée pour étayer un diagnostic d’oligodendrogliome et pour prédire la chimiosensibilité et le pronostic de ces patients 5,20. Sur la base des résultats des essais EORTC 26951 et RTOG 9402, les patients atteints de tumeurs oligodendrogliales anaplasiques à co-délétion 1p/19q ne devraient pas être traités par radiothérapie seule, mais plutôt recevoir une chimiothérapie alkylante précoce associée à une radiothérapie 20. Une approche similaire devrait être envisagée pour un sous-ensemble de patients atteints d’oligodendrogliome de grade II.
Lorsqu’un traitement médical doit être initié, l’ampleur du bénéfice thérapeutique de l’association radiothérapie plus VPC est substantielle, mais les effets toxiques sont plus importants chez les patients recevant cette association. Des essais cliniques sont en cours pour évaluer l’intérêt d’une chimiothérapie initiale seule ou d’une radiothérapie combinée plus témozolomide par rapport à une radiothérapie standard plus PCV.
Références clés
- Ostrom QT, Bauchet L, Davis FG, et al. L’épidémiologie du gliome chez l’adulte : une revue de » l’état de la science « . Neuro Oncol 2014 ; 16(7):896-913.
- Ibdaih A, Marie Y, Pierron G, et al. Deux types de pertes du chromosome 1p avec une signification opposée dans les gliomes. Ann Neurol 2005 ; 58(3):483-7.
- Jenkins R, Blair H, Ballman K, et al. Un t(1;19)(q10;p10) intervient dans les délétions combinées de 1p et 19q et prédit un meilleur pronostic des patients atteints d’oligodendrogliome. Cancer Res 2006 ; 66 : 9852-61.
- Reifenberger J, Reifenberger G, Liu L, et al. L’analyse génétique moléculaire des tumeurs oligodendrogliales montre des délétions alléliques préférentielles sur 19q et 1p. Am J Pathol 1994 ; 145:1175-90.
- Stupp R, Brada M, van den Bent M, et al. High-grade glioma : Lignes directrices de pratique clinique de l’ESMO pour le diagnostic, le traitement et le suivi. Ann Oncol 2014;25 (Suppl 3) : iii93-101.
- Louis D, Perry A, Reifenberger G. et al. La classification 2016 de l’Organisation mondiale de la santé des tumeurs du système nerveux central : un résumé. Acta Neuropathologica 2016 ; 131 : 803-20.
- Réseau de recherche Atlas du génome du cancer, Brat D, Verhaak R, et al. Analyse génomique complète et intégrative des gliomes diffus de bas grade. N Engl J Med 2015 ; 372 : 2481-98.
- Sanson M, Marie Y, Paris S, et al. La mutation du codon 132 de l’isocitrate déshydrogénase 1 est un biomarqueur pronostique important dans les gliomes. J Clin Oncol 2009 ; 27 : 4150-54.
- Bettegowda C, Agrawal N, Jiao Y, et al. Les mutations de CIC et FUBP1 contribuent à l’oligodendrogliome humain. Science 2011 ; 333(6048) : 1453-5.
- Zhao J, Ma W, Zhao H. Perte d’hétérozygotie 1p/19q et survie dans le gliome : une méta-analyse. Neuro-Oncologie 2014 ; 16 : 103-112.
- van den Bent M, Carpentier A, Brandes A, et al. La procarbazine, la lomustine et la vincristine adjuvantes améliorent la survie sans progression mais pas la survie globale dans les oligodendrogliomes et oligoastrocytomes anaplasiques nouvellement diagnostiqués : un essai de phase III randomisé de l’Organisation européenne pour la recherche et le traitement du cancer. J Clin Oncol 2006 ; 24 : 2715-22
- Cairncross G, Berkey B, Shaw E, et al. Essai de phase III de la chimiothérapie plus la radiothérapie par rapport à la radiothérapie seule pour l’oligodendrogliome anaplasique pur et mixte : Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 2006 ; 24 : 2707-14.
- Wiestler B, Capper D, Sill M, et al. Le profilage intégré de la méthylation de l’ADN et du nombre de copies identifie trois groupes de gliomes anaplasiques cliniquement et biologiquement pertinents. Acta Neuropathologica 2014 ; 128:561-71.
- Ceccarelli M, Barthel FP, Malta TM, et al. Le profilage moléculaire révèle des sous-ensembles biologiquement discrets et des voies de progression dans le gliome diffus. Cell 2016 ; 164(3) : 550-63.
- van den Bent M, Brandes A, Taphoorn M, et al. Chimiothérapie adjuvante à base de procarbazine, lomustine et vincristine dans les oligodendrogliomes anaplasiques nouvellement diagnostiqués : suivi à long terme de l’étude 26951 de l’EORTC brain tumor group. J Clin Oncol 2013 ; 31:344-50.
- Cairncross G, Wang M, Shaw E, et al. Essai de phase III de la chimioradiothérapie pour l’oligodendrogliome anaplasique : résultats à long terme du RTOG 9402. J Clin Oncol 2013 ; 31 : 337-43.
- Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus Procarbazine, CCNU, et Vincristine dans les gliomes de bas grade. N Engl J Med 2016 ; 374 : 1344-1355.
- Woehrer A, Sander P, Baberler C, et al. Détection par FISH de la codélétion 1p 19q dans les tumeurs oligodendrogliales : procédures et protocoles pour la pratique neuropathologique – une publication sous les auspices du Comité de recherche de la Confédération européenne des sociétés de neuropathologie (Euro-CNS). Clinical Neuropathology 2011 ; 30 : 47-55.
- Pinkham M, Telford N, Whitfield G, et al. FISHing Tips : Ce que chaque clinicien devrait savoir sur l’analyse 1p19q dans les gliomes en utilisant l’hybridation in situ en fluorescence. Clinical Oncology 2015 ; 27 : 445-53.
- Weller M, van den Bent M, Hopkins K, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol 2014 ; 15 : 395-403.