O princípio Franck-Condon é uma regra em espectroscopia e química quântica que explica a intensidade das transições vibronicas. As transições vibrônicas são as mudanças simultâneas nos níveis de energia eletrônica e vibracional de uma molécula devido à absorção ou emissão de um fóton da energia apropriada. O princípio afirma que durante uma transição eletrônica, uma mudança de um nível de energia vibracional para outro será mais provável de acontecer se as duas funções de ondas vibracionais se sobrepuserem mais significativamente.
Conhecimento adicional recomendado
Conteúdo
- 1 Visão geral
- 2 Desenvolvimento do princípio
- 3 Formulação mecânica quântica
- 4 metáforas Franck-Condon em espectroscopia
- 4.1 Princípio de Franck-Condon para fonemas
- 4.2 Princípio de Franck-Condon na solvação
>
- 5 Referências
- 6 Veja também
Visão geral
O princípio de Franck-Condon tem uma interpretação semiclássica bem estabelecida baseada nas contribuições originais de James Franck . As transições eletrônicas são essencialmente instantâneas em comparação com a escala de tempo dos movimentos nucleares, portanto, para que a molécula se mova para um novo nível vibracional durante a transição eletrônica, este novo nível vibracional deve ser instantaneamente compatível com as posições nucleares e o momento do nível vibracional da molécula no estado eletrônico de origem. Na imagem semiclássica das vibrações (oscilações) de um oscilador harmónico simples, as condições necessárias podem ocorrer nos pontos de viragem, onde o momento é zero.
Classicamente, o princípio Franck-Condon é a aproximação de que uma transição eletrônica é mais provável de ocorrer sem mudanças nas posições dos núcleos na entidade molecular e seu ambiente. O estado resultante é chamado de estado Franck-Condon, e a transição envolvida, uma transição vertical. A formulação mecânica quântica deste princípio é que a intensidade de uma transição vibrônica é proporcional ao quadrado da integral sobreposta entre as funções vibratórias dos dois estados que estão envolvidos na transição. – Compêndio de Terminologia Química da IUPAC, 2ª Edição (1997)
No quadro mecânico quântico, os níveis vibracionais e as funções de onda vibracionais são os dos osciladores harmónicos quânticos, ou de aproximações mais complexas à energia potencial das moléculas, como o potencial Morse. A Figura 1 ilustra o princípio de Franck-Condon para transições vibrônicas em uma molécula com funções de energia potencial morse tanto no solo quanto em estados eletrônicos excitados. Na aproximação de baixa temperatura, a molécula começa no nível v = 0 vibracional do estado eletrônico terrestre e ao absorver um fóton da energia necessária, faz uma transição para o estado eletrônico excitado. A configuração eletrônica do novo estado pode resultar em uma mudança da posição de equilíbrio dos núcleos que constituem a molécula. Na figura essa mudança nas coordenadas nucleares entre o solo e o primeiro estado excitado é rotulada como q 10. No caso mais simples de uma molécula diatômica, o eixo das coordenadas nucleares refere-se à separação internuclear. A transição vibrônica é indicada por uma seta vertical devido à suposição de coordenadas nucleares constantes durante a transição. A probabilidade da molécula poder terminar em qualquer nível vibracional particular é proporcional ao quadrado da sobreposição (vertical) das funções de onda vibracionais do estado original e final (ver seção de formulação mecânica quântica abaixo). No estado excitado eletrônico as moléculas relaxam rapidamente ao nível vibracional mais baixo (regra de Kasha), e de lá podem decair para o estado eletrônico mais baixo via emissão de fótons. O princípio Franck-Condon é aplicado igualmente à absorção e à fluorescência.
A aplicabilidade do princípio Franck-Condon tanto na absorção como na fluorescência, juntamente com a regra de Kasha leva a uma simetria de espelho aproximada mostrada na Figura 2. A estrutura vibracional das moléculas em um gás frio e esparso é mais claramente visível devido à ausência de um alargamento não homogêneo das transições individuais. As transições vibratórias são desenhadas na figura 2 como formas de linhas Lorentzianas estreitas, igualmente espaçadas. O mesmo espaçamento entre níveis vibracionais é apenas o caso do potencial parabólico de osciladores harmônicos simples, em potenciais mais realistas, como os mostrados na Figura 1, o espaçamento energético diminui com o aumento da energia vibracional. As transições eletrônicas de e para os estados vibracionais mais baixos são frequentemente referidas como transições 0-0 (zero zero zero) e têm a mesma energia tanto na absorção quanto na fluorescência.
Desenvolvimento do princípio
Num relatório publicado em 1926 pela Transactions of the Faraday Society, James Franck estava preocupado com os mecanismos das reações químicas induzidas por fótons. O mecanismo presumido era a excitação de uma molécula por um fóton seguido por uma colisão com outra molécula durante o curto período de excitação. A questão era se era possível uma molécula quebrar em fotoprodutos em uma única etapa, a absorção de um fóton, e sem uma colisão. Para que uma molécula se quebre, ela deve adquirir do fóton energia vibracional que exceda a energia de dissociação, ou seja, a energia para quebrar uma ligação química. Entretanto, como era conhecido na época, as moléculas só absorverão energia correspondente às transições quânticas permitidas e não há nível vibracional acima do nível da energia de dissociação do poço potencial. A absorção de fotões de alta energia leva a uma transição para um estado electrónico superior em vez de uma dissociação. Ao examinar quanta energia vibracional uma molécula pode adquirir quando está excitada a um nível electrónico superior, e se esta energia vibracional pode ser suficiente para quebrar imediatamente a molécula, ele desenhou três diagramas representando as possíveis mudanças na energia de ligação entre o estado electrónico mais baixo e os estados electrónicos superiores.
O diagrama I. mostra um grande enfraquecimento da ligação numa transição do estado normal n para os estados excitados a e a’. Aqui temos D > D’ e D’ > D”. Ao mesmo tempo a posição de equilíbrio dos núcleos se move com a excitação para valores maiores de r. Se passarmos da posição de equilíbrio (o mínimo de energia potencial) da curva n verticalmente para cima para uma curva no Diagrama I. as partículas terão uma energia potencial maior que D’ e voarão separadas. Neste caso temos uma mudança muito grande na energia de oscilação na excitação pela luz… – James Franck 1926
James Franck reconheceu que mudanças nos níveis vibracionais poderiam ser uma consequência da natureza instantânea da excitação para níveis de energia eletrônica mais altos e uma nova posição de equilíbrio para o potencial de interação nuclear. Edward Condon estendeu essa percepção além das fotorreações em um artigo de Revisão Física de 1926 intitulado “A Theory of Intensity Distribution in Band Systems” (Uma Teoria da Distribuição de Intensidade em Sistemas de Banda). Aqui ele formula a formulação semiclássica de uma forma bastante semelhante à sua forma moderna. A primeira referência conjunta a Franck e Condon em relação ao novo princípio aparece na mesma edição de 1926 da Physical Review em um artigo sobre a estrutura da banda de monóxido de carbono de Raymond Birge.
Formulação Mecânica Quântica
Considerar uma transição dipolo elétrica do estado vibracional inicial (ʋ) do nível eletrônico terrestre (ε), 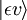 , para algum estado vibracional (ʋ’ ) de um estado eletrônico excitado (ε’),
, para algum estado vibracional (ʋ’ ) de um estado eletrônico excitado (ε’), 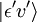 (ver notação de bra-ket). O operador do dipolo molecular μ é determinado pela carga (-e) e localizações (ri) dos elétrons, bem como as cargas (+eZj) e localizações (Rj) dos núcleos:
(ver notação de bra-ket). O operador do dipolo molecular μ é determinado pela carga (-e) e localizações (ri) dos elétrons, bem como as cargas (+eZj) e localizações (Rj) dos núcleos:
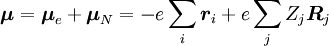
A amplitude de probabilidade P para a transição entre estes dois estados é dada por
P =ânguloângulo esquerdo {\i}psi’|direita|boldsymbol{\i} {\i}psi’^ * As funções gerais das ondas são o produto das vibrações individuais (dependendo das coordenadas espaciais dos núcleos) e do espaço electrónico e das funções das ondas de spin: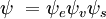
Esta separação das funções das ondas electrónicas e vibracionais é uma expressão da aproximação Born-Oppenheimer e é a suposição fundamental do princípio de Franck-Condon. A combinação destas equações leva a uma expressão da amplitude de probabilidade em termos de espaço electrónico separado, spin e contribuições vibracionais:
P =ângulo esquerdo \psi _e’ \psi _v’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ \psi _s’ (Símbolo de fivela _e + Símbolo de fivela _n)psi _e _e _v, dtau _s, d_______2621>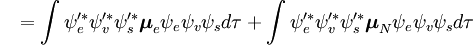
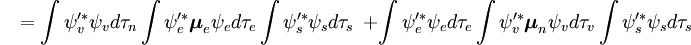
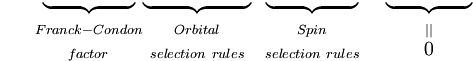
A parte independente do spin do integral inicial é aqui aproximada como um produto de dois integrais
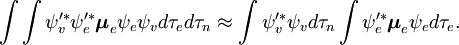
Esta factorização seria exacta se a integral 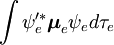 sobre as coordenadas espaciais dos elétrons não dependeria das coordenadas nucleares. Entretanto, na aproximação Born-Oppenheimer
sobre as coordenadas espaciais dos elétrons não dependeria das coordenadas nucleares. Entretanto, na aproximação Born-Oppenheimer 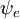 e
e 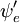 dependem (parametricamente) das coordenadas nucleares, de modo que a integral (uma chamada superfície dipolo de transição) é uma função das coordenadas nucleares. Como a dependência é normalmente bastante suave, a sua negligência (ou seja, a suposição de que a superfície dipolo de transição é independente das coordenadas nucleares) é frequentemente permitida.
dependem (parametricamente) das coordenadas nucleares, de modo que a integral (uma chamada superfície dipolo de transição) é uma função das coordenadas nucleares. Como a dependência é normalmente bastante suave, a sua negligência (ou seja, a suposição de que a superfície dipolo de transição é independente das coordenadas nucleares) é frequentemente permitida.
A primeira integral após o sinal de mais é igual a zero porque as funções de ondas eletrônicas de diferentes estados são ortogonais. O restante é o produto de três integrais. A primeira integral é a integral de sobreposição vibracional, também chamada fator Franck-Condon. Os dois integrais restantes que contribuem para a amplitude de probabilidade determinam as regras de seleção eletrônica espacial e de spin.
O princípio Franck-Condon é uma declaração sobre transições vibracionais permitidas entre dois estados eletrônicos diferentes, outras regras de seleção mecânica quântica podem diminuir a probabilidade de uma transição ou proibi-la completamente. As regras de seleção rotacional têm sido negligenciadas na derivação acima. Contribuições rotacionais podem ser observadas nos espectros de gases, mas são fortemente suprimidas em líquidos e sólidos.
Deve ficar claro que a formulação mecânica quântica do princípio de Franck-Condon é o resultado de uma série de aproximações, principalmente a suposição de transição dipolo elétrica e a aproximação Born-Oppenheimer. As transições eletrônicas de dipolo magnético fraco e quadrupolo elétrico, juntamente com a validade incompleta da fatorização da função de onda total em funções de ondas nucleares, espaciais eletrônicas e de ondas de spin significa que as regras de seleção, incluindo o fator Franck-Condon, não são estritamente observadas. Para qualquer transição, o valor de P é determinado por todas as regras de seleção, entretanto a seleção de spin é o maior contribuinte, seguida pelas regras de seleção eletrônica. O factor Franck-Condon apenas modula de forma fraca a intensidade das transições, ou seja, contribui com um factor na ordem de 1 para a intensidade das bandas cuja ordem de grandeza é determinada pelas outras regras de selecção. A tabela abaixo fornece a gama de coeficientes de extinção para as combinações possíveis de regras de seleção de giros e orbitais permitidos e proibidos.
>
103 a 105
>
100 a 103
10-5 a 100
Metáforas de Franck-Condon em espectroscopia
O princípio de Franck-Condon, na sua forma canónica, aplica-se apenas a alterações nos níveis vibracionais de uma molécula no decurso de uma alteração nos níveis electrónicos, quer por absorção, quer por emissão de um fotão. A intuição física deste princípio é ancorada pela ideia de que as coordenadas nucleares dos átomos que constituem a molécula não têm tempo para mudar durante o muito breve período de tempo envolvido numa transição electrónica. No entanto, esta intuição física pode ser, e é de fato, rotineiramente estendida a interações entre moléculas absorventes ou emissoras de luz (cromóforos) e seu ambiente. As metáforas de Franck-Condon são apropriadas porque as moléculas frequentemente interagem fortemente com as moléculas circundantes, particularmente em líquidos e sólidos, e essas interações modificam as coordenadas nucleares do cromóforo de maneiras muito análogas às vibrações moleculares consideradas pelo princípio de Franck-Condon.
Princípio Franck-Condon para os fonos
A analogia Franck-Condon mais próxima é devida à interação dos fonos -quanta de vibrações da malha- com as transições eletrônicas dos cromóforos embutidas como impurezas da malha. Nesta situação, as transições para níveis electrónicos mais elevados podem ocorrer quando a energia do fotão corresponde à energia de transição puramente electrónica ou à energia de transição puramente electrónica mais a energia de um ou mais fotões de malha. Na aproximação a baixa temperatura, a emissão passa do nível de zero-fão do estado excitado para o nível de zero-fão do estado do solo ou para níveis de fontões mais altos do estado do solo. Tal como no princípio de Franck-Condon, a probabilidade de transições envolvendo fonões é determinada pela sobreposição das funções das ondas de fonões nos níveis de energia inicial e final. Para o princípio de Franck-Condon aplicado às transições fonônicas, a etiqueta do eixo horizontal da Figura 1 é substituída na Figura 6 pela coordenada configurável para um modo normal. A energia potencial qi do modo treliça na Figura 6 é representada como a de um oscilador harmônico, e o espaçamento entre os níveis de fono (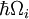 )é determinado pelos parâmetros da treliça. Como a energia dos fonões simples é geralmente bastante pequena, as transições de zero ou de poucas colunas só podem ser observadas a temperaturas inferiores a cerca de 40 kelvins.
)é determinado pelos parâmetros da treliça. Como a energia dos fonões simples é geralmente bastante pequena, as transições de zero ou de poucas colunas só podem ser observadas a temperaturas inferiores a cerca de 40 kelvins.
Ver linha de zeros e banda lateral de fonões para mais detalhes e referências.
Princípio de Franck-Condon na solvação
Considerações de Franck-Condon também podem ser aplicadas às transições eletrônicas dos cromóforos dissolvidos em líquidos. Neste uso da metáfora Franck-Condon, os níveis vibracionais dos cromóforos, assim como as interações dos cromóforos com os fonos no líquido, continuam a contribuir para a estrutura dos espectros de absorção e emissão, mas estes efeitos são considerados separada e independentemente.
Cromóforos cercados por moléculas de solventes. Estas moléculas circundantes podem interagir com os cromóforos, particularmente se as moléculas de solvente forem polares. Esta associação entre o solvente e o soluto é referida como solvente e é uma interacção estabilizadora, ou seja, as moléculas de solvente podem mover-se e rodar até que a energia da interacção seja minimizada. A interação em si envolve forças eletrostáticas e van der Waals e também pode incluir ligações de hidrogênio. Os princípios de Franck-Condon podem ser aplicados quando as interações entre o cromóforo e as moléculas de solvente circundantes são diferentes no solo e no estado eletrônico excitado. Esta mudança na interação pode ter origem, por exemplo, devido a diferentes momentos dipolares nestes dois estados. Se o cromóforo começa no seu estado de terra e está perto do equilíbrio com as moléculas de solvente circundantes e depois absorve um fotão que o leva ao estado excitado, a sua interacção com o solvente estará longe do equilíbrio no estado excitado. Este efeito é análogo ao princípio original de Franck-Condon: a transição electrónica é muito rápida em comparação com o movimento dos núcleos – o rearranjo das moléculas de solvente no caso da solvência -. Falamos agora de uma transição vertical, mas agora a coordenada horizontal é o espaço de interação soluto-soluto-soluto-soluto. Este eixo de coordenadas é frequentemente rotulado como “Coordenada de Solvação” e representa, de forma algo abstracta, todas as dimensões relevantes do movimento de todas as moléculas de solvente em interacção.
No princípio original de Franck-Condon, após a transição electrónica, as moléculas que acabam em estados vibracionais mais elevados começam imediatamente a relaxar para o estado vibracional mais baixo. No caso da solvência, as moléculas de solvente tentarão imediatamente reorganizar-se de forma a minimizar a energia de interacção. A taxa de relaxamento do solvente depende da viscosidade do solvente. Assumindo que o tempo de relaxamento do solvente é curto em comparação com a vida útil do estado excitado eletrônico, a emissão será do estado de menor energia do solvente do estado excitado eletrônico. Para solventes de pequenas moléculas, como água ou metanol à temperatura ambiente, o tempo de relaxamento do solvente é da ordem de algumas dezenas de picossegundos, enquanto a vida útil do estado excitado por cromóforo varia de alguns picossegundos a alguns nanossegundos. Imediatamente após a transição para o estado eletrônico terrestre, as moléculas de solvente também devem se reorganizar para acomodar a nova configuração eletrônica do cromóforo. A figura 7 ilustra o princípio de Franck-Condon aplicado à solvência. Quando a solução é iluminada pela luz correspondente à energia de transição eletrônica, alguns dos cromóforos se moverão para o estado excitado. Dentro deste grupo de cromóforos haverá uma distribuição estatística das energias de interacção solvent-chromophore, representada na figura por uma função de distribuição Gaussiana. A interação solvent-chromophore é desenhada como um potencial parabólico em ambos os estados eletrônicos. Como a transição eletrônica é essencialmente instantânea na escala temporal do movimento do solvente (seta vertical), a coleção de cromóforos de estado excitado está imediatamente longe do equilíbrio. O rearranjo das moléculas de solvente de acordo com a nova curva de energia potencial é representado pelas setas curvas na Figura 7. Note que enquanto as transições eletrônicas são quantizadas, a energia de interação cromóforo-solvente é tratada como um continuum clássico devido ao grande número de moléculas envolvidas. Embora a emissão seja descrita como ocorrendo a partir do mínimo do potencial de interação cromóforo-solvente do estado excitado, uma emissão significativa pode ocorrer antes do equilíbrio ser alcançado quando a viscosidade do solvente é alta ou a vida útil do estado excitado é curta. A diferença de energia entre os fótons absorvidos e emitidos, descrita na Figura 7, é a contribuição da solvação para o deslocamento de Stokes.
Links de periódicos podem requerer assinatura.
- Franck, J. (1926). “Processos elementares de reações fotoquímicas”. Transações da Sociedade Faraday 21: 536-542. Link
- Condon, E. (1926). “A theory of intensity distribution in band systems (Meeting abstract)”. Revisão Física 27: 640.
- Condon, E. (1926). “Uma teoria da distribuição de intensidade em sistemas de banda”. Revisão Física 28: 1182-1201. Link
- Condon, E. (1928). “Movimentos nucleares associados a transições de elétrons em moléculas diatômicas”. Revisão Física 32: 858-872. Link
- Birge, R. T. (1926). “The band spectra of carbon monoxide”. Revisão Física 28: 1157-1181. Link
- Noyes, W. A. (1933). “The correlation of spectroscopy and photochemistry”. Revisões de Física Moderna 5: 280-287. Link
- Coolidge, A. S, James, H. M. e Present, R. D. (1936). “A study of the Franck-Condon Principle” (Um estudo do Princípio de Franck-Condon). Journal of Chemical Physics 4: 193-211. Link
- Herzberg, Gerhard (1971). Os espectros e as estruturas dos radicais livres simples. Nova York: Dover. ISBN 0-486-65821-X.
- Harris, Daniel C.; Michael D. Bertolucci (1978). Simetria e espectroscopia. Nova York: Dover. ISBN 0-486-66144-X.
- Bernath, Peter F. (1995). Spectra of Atoms and Molecules (Tópicos em Química Física). Oxford: Oxford University Press. ISBN 0-19-507598-6.
- Atkins, P. W.; R. S. Frieman (1999). Mecânica Quântica Molecular. Oxford: Oxford University Press. ISBN 0-19-855947-X.
>
>
Ver também
- Aproximação Born-Oppenheimer
- Transição electrónica molecular
- Ultravioleta.espectroscopia visível
- Oscilador harmónico quântico
- Potencial morse
- Acoplamento vibrónico
- Linha de zeros e banda lateral de zeros
- Aproximação súbita
Categorias: Química quântica | Espectroscopia | Física molecular