Principiul lui Franck-Condon este o regulă în spectroscopie și în chimia cuantică care explică intensitatea tranzițiilor vibronice. Tranzițiile vibronice sunt modificările simultane ale nivelurilor energetice electronice și vibraționale ale unei molecule ca urmare a absorbției sau emiterii unui foton de energie corespunzătoare. Principiul afirmă că, în timpul unei tranziții electronice, schimbarea de la un nivel energetic vibrațional la altul va fi mai probabilă dacă cele două funcții de undă vibraționale se suprapun în mod semnificativ.
Cunoștințe suplimentare recomandate
Contenit
- 1 Prezentare generală
- 2 Dezvoltarea principiului
- 3 Formularea mecanică cuantică
- 4 Metaforele Franck-Condon în spectroscopie
- 4.1 Principiul Franck-Condon pentru fononi
- 4.2 Principiul Franck-Condon în solvatare
- 5 Referințe
- 6 Vezi și
Prezentare generală
Principiul Franck-Condon are o interpretare semiclasică bine stabilită, bazată pe contribuțiile originale ale lui James Franck . Tranzițiile electronice sunt în esență instantanee în comparație cu scara de timp a mișcărilor nucleare, prin urmare, dacă molecula trebuie să se deplaseze la un nou nivel vibrațional în timpul tranziției electronice, acest nou nivel vibrațional trebuie să fie instantaneu compatibil cu pozițiile și momentele nucleare ale nivelului vibrațional al moleculei în starea electronică de origine. În imaginea semiclasică a vibrațiilor (oscilațiilor) unui oscilator armonic simplu, condițiile necesare pot avea loc în punctele de cotitură, unde momentul este zero.
Din punct de vedere clasic, principiul Franck-Condon reprezintă aproximația conform căreia o tranziție electronică are cea mai mare probabilitate de a se produce fără modificări ale pozițiilor nucleelor din entitatea moleculară și din mediul acesteia. Starea rezultată se numește o stare Franck-Condon, iar tranziția implicată, o tranziție verticală. Formularea mecanică cuantică a acestui principiu este că intensitatea unei tranziții vibronice este proporțională cu pătratul integralei de suprapunere dintre funcțiile de undă vibraționale ale celor două stări care sunt implicate în tranziție. – IUPAC Compendium of Chemical Terminology, ediția a 2-a (1997)
În imaginea mecanică cuantică, nivelurile vibraționale și funcțiile de undă vibraționale sunt cele ale oscilatorilor armonici cuantici, sau ale unor aproximări mai complexe ale energiei potențiale a moleculelor, cum ar fi potențialul Morse. Figura 1 ilustrează principiul Franck-Condon pentru tranzițiile vibronice într-o moleculă cu funcții de energie potențială de tip Morse atât în starea electronică fundamentală, cât și în cea excitată. În aproximația la temperaturi scăzute, molecula începe în nivelul vibrațional v = 0 al stării electronice fundamentale și, la absorbția unui foton cu energia necesară, face o tranziție la starea electronică excitată. Configurația electronică a noii stări poate duce la o deplasare a poziției de echilibru a nucleelor care constituie molecula. În figură, această deplasare a coordonatelor nucleare între starea fundamentală și prima stare excitată este marcată cu q 10. În cazul cel mai simplu al unei molecule diatomice, axa coordonatelor nucleare se referă la separarea internucleară. Tranziția vibronică este indicată printr-o săgeată verticală datorită presupunerii unor coordonate nucleare constante în timpul tranziției. Probabilitatea ca molecula să ajungă într-un anumit nivel vibrațional este proporțională cu pătratul suprapunerii (verticale) a funcțiilor de undă vibraționale ale stării inițiale și finale (a se vedea secțiunea de formulare mecanică cuantică de mai jos). În starea electronică excitată, moleculele se relaxează rapid până la cel mai scăzut nivel vibrațional (regula lui Kasha), iar de acolo se pot dezintegra până la cea mai scăzută stare electronică prin emisie de fotoni. Principiul Franck-Condon se aplică în egală măsură la absorbție și la fluorescență.
Aplicarea principiului Franck-Condon atât la absorbție, cât și la fluorescență, împreună cu regula lui Kasha conduce la o simetrie în oglindă aproximativă prezentată în figura 2. Structura vibrațională a moleculelor într-un gaz rece și rarefiat este cel mai clar vizibilă datorită absenței lărgirii neomogene a tranzițiilor individuale. Tranzițiile vibronice sunt desenate în figura 2 ca forme de linii lorentziene înguste și egal distanțate. Spațierea egală între nivelurile vibraționale este doar cazul pentru potențialul parabolic al oscilatorilor armonici simpli, în cazul unor potențiale mai realiste, cum ar fi cele prezentate în figura 1, spațierea energetică scade odată cu creșterea energiei vibraționale. Tranzițiile electronice către și dinspre cele mai joase stări vibraționale sunt adesea denumite tranziții 0-0 (zero zero zero) și au aceeași energie atât în absorbție, cât și în fluorescență.
Dezvoltarea principiului
Într-un raport publicat în 1926 de către Transactions of the Faraday Society, James Franck a fost preocupat de mecanismele reacțiilor chimice induse de fotoni. Mecanismul presupus a fost excitarea unei molecule de către un foton urmată de o coliziune cu o altă moleculă în timpul scurtei perioade de excitație. Întrebarea era dacă este posibil ca o moleculă să se descompună în fotoproduse într-o singură etapă, absorbția unui foton, și fără o coliziune. Pentru ca o moleculă să se descompună, aceasta trebuie să dobândească de la foton o energie vibrațională care să depășească energia de disociere, adică energia necesară pentru a rupe o legătură chimică. Cu toate acestea, așa cum se știa la vremea respectivă, moleculele vor absorbi numai energia corespunzătoare tranzițiilor cuantice permise și nu există niciun nivel vibrațional peste nivelul de energie de disociere al puțului potențial. Absorbția fotonilor de mare energie duce la o tranziție către o stare electronică superioară în locul disocierii. Examinând câtă energie de vibrație ar putea dobândi o moleculă atunci când este excitată la un nivel electronic superior și dacă această energie de vibrație ar putea fi suficientă pentru a disocia imediat molecula, el a desenat trei diagrame reprezentând posibilele modificări ale energiei de legătură între starea electronică cea mai joasă și stările electronice superioare.
Diagrama I. arată o mare slăbire a legăturii la o tranziție de la starea normală n la stările excitate a și a’. Aici avem D > D’ și D’ > D”. În același timp, poziția de echilibru a nucleelor se deplasează odată cu excitarea la valori mai mari ale lui r. Dacă trecem de la poziția de echilibru (minimul de energie potențială) a curbei n pe verticală în sus la curbele a din diagrama I. particulele vor avea o energie potențială mai mare decât D’ și se vor despărți în zbor. În acest caz avem o schimbare foarte mare a energiei de oscilație la excitarea prin lumină… – James Franck 1926
James Franck a recunoscut că modificările nivelurilor de vibrație ar putea fi o consecință a caracterului instantaneu al excitării la niveluri superioare de energie electronică și o nouă poziție de echilibru pentru potențialul de interacțiune nucleară. Edward Condon a extins această perspectivă dincolo de fotoreacțiile din 1926 într-un articol din Physical Review intitulat „A Theory of Intensity Distribution in Band Systems”. Aici el formulează formularea semiclasică într-o manieră destul de asemănătoare cu forma sa modernă. Prima referire comună atât la Franck, cât și la Condon în ceea ce privește noul principiu apare în același număr din 1926 al revistei Physical Review, într-un articol despre structura de bandă a monoxidului de carbon, scris de Raymond Birge.
Formulare de mecanică cuantică
Considerăm o tranziție de dipol electric de la starea de vibrație inițială (ʋ) a nivelului electronic de bază (ε), 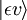 , la o anumită stare de vibrație (ʋ’ ) a unei stări electronice excitate (ε’),
, la o anumită stare de vibrație (ʋ’ ) a unei stări electronice excitate (ε’), 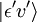 (vezi notația bra-ket). Operatorul de dipol molecular μ este determinat de sarcina (-e) și locațiile (ri) electronilor, precum și de sarcinile (+eZj) și locațiile (Rj) nucleelor:
(vezi notația bra-ket). Operatorul de dipol molecular μ este determinat de sarcina (-e) și locațiile (ri) electronilor, precum și de sarcinile (+eZj) și locațiile (Rj) nucleelor:
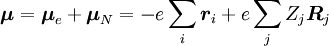
Amplitudinea probabilității P pentru tranziția între aceste două stări este dată de
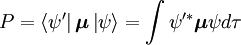
Unde 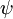 și
și 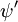 sunt, respectiv, funcțiile de undă globale ale stării inițiale și finale. Funcțiile de undă globale sunt produsul funcțiilor de undă individuale vibraționale (în funcție de coordonatele spațiale ale nucleelor) și electronice de spațiu și de spin:
sunt, respectiv, funcțiile de undă globale ale stării inițiale și finale. Funcțiile de undă globale sunt produsul funcțiilor de undă individuale vibraționale (în funcție de coordonatele spațiale ale nucleelor) și electronice de spațiu și de spin:
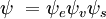
Această separare a funcțiilor de undă electronice și vibraționale este o expresie a aproximației Born-Oppenheimer și reprezintă ipoteza fundamentală a principiului Franck-Condon. Combinarea acestor ecuații conduce la o expresie pentru amplitudinea de probabilitate în termeni de spațiu electronic separat, contribuții de spin și vibraționale:
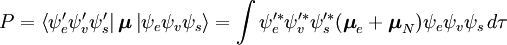
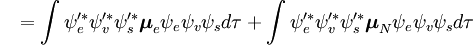 {\color{White}P}=\int { \psi _v’^ * } \psi _v d\tau_n \int { \psi _e’^ * * } \boldsymbol{\mu} _e \psi _e d\tau_e \int { \psi _s’^’* } \psi _s d\tau_s \ \ \ + \int { \psi _e’^ * } \psi _e d\tau_e \int { \psi _v’^ * * } \boldsymbol{\mu} _n \psi _v d\tau_v \int { \psi _s’^ * } \psi _s d\tau_s
{\color{White}P}=\int { \psi _v’^ * } \psi _v d\tau_n \int { \psi _e’^ * * } \boldsymbol{\mu} _e \psi _e d\tau_e \int { \psi _s’^’* } \psi _s d\tau_s \ \ \ + \int { \psi _e’^ * } \psi _e d\tau_e \int { \psi _v’^ * * } \boldsymbol{\mu} _n \psi _v d\tau_v \int { \psi _s’^ * } \psi _s d\tau_s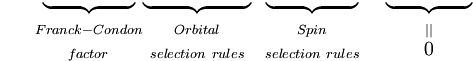
Partea independentă de spin a integralei inițiale este aici aproximată ca un produs a două integrale
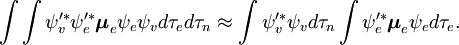
Această factorizare ar fi exactă dacă integrala 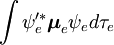 asupra coordonatelor spațiale ale electronilor nu ar depinde de coordonatele nucleare. Cu toate acestea, în aproximația Born-Oppenheimer
asupra coordonatelor spațiale ale electronilor nu ar depinde de coordonatele nucleare. Cu toate acestea, în aproximația Born-Oppenheimer 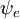 și
și 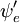 depind (parametric) de coordonatele nucleare, astfel încât integrala (o așa-numită suprafață de dipol de tranziție) este o funcție a coordonatelor nucleare. Deoarece dependența este de obicei destul de lină, neglijarea acesteia (adică presupunerea că suprafața dipolului de tranziție este independentă de coordonatele nucleare) este adesea permisă.
depind (parametric) de coordonatele nucleare, astfel încât integrala (o așa-numită suprafață de dipol de tranziție) este o funcție a coordonatelor nucleare. Deoarece dependența este de obicei destul de lină, neglijarea acesteia (adică presupunerea că suprafața dipolului de tranziție este independentă de coordonatele nucleare) este adesea permisă.
Prima integrală după semnul plus este egală cu zero deoarece funcțiile de undă electronice ale diferitelor stări sunt ortogonale. Restul este produsul a trei integrale. Prima integrală este integrala de suprapunere vibrațională, numită și factorul Franck-Condon. Celelalte două integrale care contribuie la amplitudinea probabilității determină regulile de selecție electronică spațială și de spin.
Principiul Franck-Condon este o afirmație privind tranzițiile vibraționale permise între două stări electronice diferite, alte reguli de selecție mecanică cuantică pot scădea probabilitatea unei tranziții sau o pot interzice cu totul. Regulile de selecție rotațională au fost neglijate în derivarea de mai sus. Contribuțiile rotaționale pot fi observate în spectrele gazelor, dar sunt puternic suprimate în lichide și solide.
Ar trebui să fie clar că formularea mecanică cuantică a principiului Franck-Condon este rezultatul unei serii de aproximări, în principal ipoteza tranziției de dipol electric și aproximația Born-Oppenheimer. Tranzițiile electronice de dipol magnetic și de cuadripol electric mai slabe, împreună cu valabilitatea incompletă a factorizării funcției de undă totale în funcții de undă nucleare, electronice spațiale și de spin, înseamnă că regulile de selecție, inclusiv factorul Franck-Condon, nu sunt respectate cu strictețe. Pentru orice tranziție dată, valoarea lui P este determinată de toate regulile de selecție, însă selecția de spin este cea care contribuie cel mai mult, urmată de regulile de selecție electronică. Factorul Franck-Condon modulează doar slab intensitatea tranzițiilor, și anume, contribuie cu un factor de ordinul 1 la intensitatea benzilor al căror ordin de mărime este determinat de celelalte reguli de selecție. Tabelul de mai jos prezintă gama de coeficienți de extincție pentru combinațiile posibile de reguli de selecție a spinului și orbitalului permise și interzise.
103 până la 105
100 până la 103
10-5 până la 100
Metafore Franck-Condon în spectroscopie
Principiul Franck-Condon, în forma sa canonică, se aplică numai modificărilor nivelurilor vibraționale ale unei molecule în cursul unei modificări a nivelurilor electronice, fie prin absorbția, fie prin emisia unui foton. Intuiția fizică a acestui principiu este ancorată de ideea că coordonatele nucleare ale atomilor care constituie molecula nu au timp să se modifice în timpul foarte scurt implicat într-o tranziție electronică. Cu toate acestea, această intuiție fizică poate fi și este, de fapt, extinsă în mod obișnuit la interacțiunile dintre moleculele care absorb sau emit lumină (cromofori) și mediul lor. Metaforele Franck-Condon sunt adecvate deoarece moleculele interacționează adesea puternic cu moleculele din jur, în special în lichide și solide, iar aceste interacțiuni modifică coordonatele nucleare ale cromoforului în moduri care sunt foarte asemănătoare cu vibrațiile moleculare luate în considerare de principiul Franck-Condon.
Principiul Franck-Condon pentru fononi
Cea mai apropiată analogie Franck-Condon se datorează interacțiunii fononilor -cuante de vibrații ale rețelei- cu tranzițiile electronice ale cromoforilor încorporați ca impurități în rețea. În această situație, tranzițiile către niveluri electronice superioare pot avea loc atunci când energia fotonului corespunde energiei de tranziție pur electronică sau energiei de tranziție pur electronică plus energia unuia sau mai multor fononi de rețea. În apropierea temperaturii joase, emisia are loc de la nivelul de fonon zero al stării excitate la nivelul de fonon zero al stării fundamentale sau la niveluri de fonon superioare ale stării fundamentale. La fel ca în cazul principiului Franck-Condon, probabilitatea tranzițiilor care implică fononi este determinată de suprapunerea funcțiilor de undă ale fononilor la nivelurile energetice inițiale și finale. Pentru principiul Franck-Condon aplicat la tranzițiile fononice, eticheta axei orizontale din figura 1 este înlocuită în figura 6 cu coordonata configurațională pentru un mod normal. Energia potențială a modului de rețea qi din figura 6 este reprezentată ca fiind cea a unui oscilator armonic, iar distanța dintre nivelurile fononice (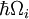 )este determinată de parametrii rețelei. Deoarece energia fononilor unici este, în general, destul de mică, tranzițiile cu zero sau cu puțini fononi pot fi observate numai la temperaturi mai mici de aproximativ 40 kelvins.
)este determinată de parametrii rețelei. Deoarece energia fononilor unici este, în general, destul de mică, tranzițiile cu zero sau cu puțini fononi pot fi observate numai la temperaturi mai mici de aproximativ 40 kelvins.
Vezi Linia de fononi zero și banda laterală de fononi pentru mai multe detalii și referințe.
Principiul Franck-Condon în solvatare
Considerațiile lui Franck-Condon pot fi aplicate și la tranzițiile electronice ale cromoforilor dizolvați în lichide. În această utilizare a metaforei Franck-Condon, nivelurile vibraționale ale cromoforilor, precum și interacțiunile cromoforilor cu fononii din lichid, continuă să contribuie la structura spectrelor de absorbție și emisie, dar aceste efecte sunt considerate separat și independent.
Considerăm cromoforii înconjurați de molecule de solvent. Aceste molecule din jur pot interacționa cu cromoforii, în special dacă moleculele de solvent sunt polare. Această asociere dintre solvent și solut se numește solvatare și este o interacțiune stabilizatoare, adică moleculele de solvent se pot mișca și roti până când energia interacțiunii este minimizată. Interacțiunea în sine implică forțe electrostatice și forțe van der Waals și poate include, de asemenea, legături de hidrogen. Principiile Franck-Condon pot fi aplicate atunci când interacțiunile dintre cromofor și moleculele de solvent din jur sunt diferite în starea electronică fundamentală și în cea excitată. Această modificare a interacțiunii poate proveni, de exemplu, din cauza momentelor dipolare diferite în aceste două stări. Dacă cromoforul începe în starea fundamentală și este aproape de echilibru cu moleculele de solvent din jur și apoi absoarbe un foton care îl duce în starea excitată, interacțiunea sa cu solventul va fi departe de echilibru în starea excitată. Acest efect este analog cu principiul original Franck-Condon: tranziția electronică este foarte rapidă în comparație cu mișcarea nucleelor – rearanjarea moleculelor de solvent în cazul solvației -. Vorbim acum de o tranziție verticală, dar acum coordonata orizontală este spațiul de interacțiune solvent-solut. Această axă de coordonate este adesea etichetată drept „coordonate de solvatare” și reprezintă, oarecum abstract, toate dimensiunile relevante ale mișcării tuturor moleculelor de solvent care interacționează.
În principiul Franck-Condon original, după tranziția electronică, moleculele care ajung în stări vibraționale superioare încep imediat să se relaxeze la starea vibrațională cea mai joasă. În cazul solvării, moleculele de solvent vor încerca imediat să se rearanjeze pentru a minimiza energia de interacțiune. Rata de relaxare a solventului depinde de vâscozitatea solventului. Presupunând că timpul de relaxare a solventului este scurt în comparație cu durata de viață a stării electronice excitate, emisia se va produce din cea mai joasă stare de energie a solventului din starea electronică excitată. Pentru solvenții cu molecule mici, cum ar fi apa sau metanolul la temperatura ambiantă, timpul de relaxare a solventului este de ordinul câtorva zeci de picosecunde, în timp ce durata de viață a stării excitate a cromoforului variază de la câteva picosecunde la câteva nanosecunde. Imediat după tranziția la starea electronică fundamentală, moleculele de solvent trebuie, de asemenea, să se rearanjeze pentru a se adapta la noua configurație electronică a cromoforului. Figura 7 ilustrează principiul Franck-Condon aplicat la solvatare. Atunci când soluția este iluminată cu o lumină corespunzătoare energiei electronice de tranziție, unii dintre cromofori se vor muta în starea excitată. În cadrul acestui grup de cromofori va exista o distribuție statistică a energiilor de interacțiune solvent-cromofor, reprezentată în figură printr-o funcție de distribuție gaussiană. Interacțiunea solvent-cromofor este desenată sub forma unui potențial parabolic în ambele stări electronice. Deoarece tranziția electronică este practic instantanee pe scara de timp a mișcării solventului (săgeata verticală), colecția de cromofori în stare excitată este imediat departe de echilibru. Rearanjarea moleculelor de solvent în conformitate cu noua curbă de energie potențială este reprezentată de săgețile curbe din figura 7. Rețineți că, în timp ce tranzițiile electronice sunt cuantificate, energia de interacțiune cromofor-solvent este tratată ca un continuum clasic din cauza numărului mare de molecule implicate. Deși emisia este reprezentată ca având loc de la minimul potențialului de interacțiune cromofor-solvent în stare excitată, o emisie semnificativă poate avea loc înainte de atingerea echilibrului atunci când vâscozitatea solventului este mare sau durata de viață a stării excitate este scurtă. Diferența de energie dintre fotonii absorbiți și cei emiși, descrisă în figura 7, reprezintă contribuția de solvatare la deplasarea Stokes.
Legături de jurnal pot necesita abonament.
- Franck, J. (1926). „Procesele elementare ale reacțiilor fotochimice”. Transactions of the Faraday Society 21: 536-542. Legătură
- Condon, E. (1926). „A theory of intensity distribution in band systems (Rezumat de ședință)”. Physical Review 27: 640.
- Condon, E. (1926). „A theory of intensity distribution in band systems”. Physical Review 28: 1182-1201. Link
- Condon, E. (1928). „Nuclear motions associated with electron transitions in diatomic molecules”. Physical Review 32: 858-872. Link
- Birge, R. T. (1926). „The band spectra of carbon monoxide”. Physical Review 28: 1157-1181. Link
- Noyes, W. A. (1933). „The correlation of spectroscopy and photochemistry” (Corelația dintre spectroscopie și fotochimie). Reviews of Modern Physics 5: 280-287. Link
- Coolidge, A. S, James, H. M. și Present, R. D. (1936). „A study of the Franck-Condon Principle”. Journal of Chemical Physics 4: 193-211. Link
- Herzberg, Gerhard (1971). The spectra and structures of simple free radicals. New York: Dover. ISBN 0-486-65821-X.
- Harris, Daniel C.; Michael D. Bertolucci (1978). Simetrie și spectroscopie. New York: Dover. ISBN 0-486-66144-X.
- Bernath, Peter F. (1995). Spectrele atomilor și moleculelor (Topics in Physical Chemistry). Oxford: Oxford University Press. ISBN 0-19-507598-6.
- Atkins, P. W.; R. S. Frieman (1999). Molecular Quantum Mechanics. Oxford: Oxford University Press. ISBN 0-19-855947-X.
Vezi și
- Aproximarea Born-Oppenheimer
- Tranziția electronică moleculară
- Tranziția electronică moleculară
- Ultraviolete-vizibil
- Oscilator armonic cuantic
- Potențial Morse
- Cuplaj vibronic
- Linie de fonon zero și bandă laterală de fonon
- Aproximare bruscă
Categorii: Chimie cuantică | Spectroscopie | Fizică moleculară
.