Franck-Condon-princippet er en regel inden for spektroskopi og kvantekemi, der forklarer intensiteten af vibroniske overgange. Vibroniske overgange er de samtidige ændringer i et molekyls elektroniske og vibrationsmæssige energiniveauer som følge af absorption eller emission af en foton med den relevante energi. Princippet siger, at under en elektronisk overgang er der større sandsynlighed for, at der sker en ændring fra et vibrationsenerginiveau til et andet, hvis de to vibrationsbølgefunktioner overlapper hinanden i større omfang.
Tidligere anbefalet viden
Indhold
- 1 Oversigt
- 2 Udvikling af princippet
- 3 Kvantemekanisk formulering
- 4 Franck-Condon-metaforer i spektroskopi
- 4.1 Franck-Condon-princippet for fononer
- 4.2 Franck-Condon-princippet i solvering
- 5 Referencer
- 6 Se også
Oversigt
Det Franck-Condon-princippet har en veletableret semiklassisk fortolkning baseret på de oprindelige bidrag af James Franck . Elektroniske overgange er i det væsentlige øjeblikkelige sammenlignet med tidsskalaen for kernebevægelser, og hvis molekylet derfor skal bevæge sig til et nyt vibrationsniveau under den elektroniske overgang, skal dette nye vibrationsniveau være øjeblikkeligt kompatibelt med de nukleare positioner og momenta for molekylets vibrationsniveau i den oprindelige elektroniske tilstand. I det semiklassiske billede af vibrationer (svingninger) af en simpel harmonisk oscillator kan de nødvendige betingelser opstå ved vendepunkterne, hvor impulsen er nul.
Klassisk set er Franck-Condon-princippet den tilnærmelse, at en elektronisk overgang med størst sandsynlighed finder sted uden ændringer i kernes positioner i den molekylære enhed og dens omgivelser. Den resulterende tilstand kaldes en Franck-Condon-tilstand, og den pågældende overgang en vertikal overgang. Den kvantemekaniske formulering af dette princip er, at intensiteten af en vibronisk overgang er proportional med kvadratet på overlapningsintegralet mellem de vibrationelle bølgefunktioner i de to tilstande, der er involveret i overgangen. – IUPAC Compendium of Chemical Terminology, 2nd Edition (1997)
I det kvantemekaniske billede er vibrationsniveauerne og vibrationsbølgefunktionerne dem af kvanteharmoniske oscillatorer eller af mere komplekse tilnærmelser til molekylernes potentielle energi, som f.eks. morsepotentialet. Figur 1 illustrerer Franck-Condon-princippet for vibroniske overgange i et molekyle med Morse-lignende potentielle energifunktioner i både grundtilstande og exciterede elektroniske tilstande. I lavtemperaturapproksimationen starter molekylet i v = 0-vibrationsniveauet i den elektroniske grundtilstand, og når det absorberer en foton med den nødvendige energi, foretager det en overgang til den exciterede elektroniske tilstand. Elektronkonfigurationen i den nye tilstand kan resultere i en forskydning af ligevægtspositionen for de kerner, der udgør molekylet. I figuren er denne forskydning i kernekoordinaterne mellem grundtilstanden og den første exciterede tilstand betegnet som q 10. I det enkleste tilfælde af et toatomigt molekyle henviser kernekoordinaternes akse til den indbyrdes kerneadskillelse. Den vibroniske overgang er angivet med en lodret pil, da det antages, at kernekoordinaterne er konstante under overgangen. Sandsynligheden for, at molekylet kan ende i et bestemt vibrationsniveau, er proportional med kvadratet på det (lodrette) overlap af de vibrationsmæssige bølgefunktioner for den oprindelige og den endelige tilstand (se afsnittet om kvantemekanisk formulering nedenfor). I den elektroniske exciterede tilstand slapper molekylerne hurtigt af til det laveste vibrationsniveau (Kasha’s regel), og derfra kan de henfalde til den laveste elektroniske tilstand via fotonemission. Franck-Condon-princippet gælder både for absorption og fluorescens.
Anvendeligheden af Franck-Condon-princippet i både absorption og fluorescens fører sammen med Kasha’s regel til en tilnærmet spejlsymmetri, der er vist i figur 2. Molekylers vibrationsstruktur i en kold, sparsom gas er tydeligst synlig på grund af fraværet af inhomogen udvidelse af de enkelte overgange. Vibroniske overgange er i figur 2 tegnet som smalle, lige store Lorentzian-linjer med lige store afstande. Lige afstand mellem vibrationsniveauer er kun tilfældet for det paraboliske potentiale for simple harmoniske oscillatorer, i mere realistiske potentialer, som f.eks. dem, der er vist i figur 1, falder energiafstanden med stigende vibrationsenergi. Elektroniske overgange til og fra de laveste vibrationstilstande kaldes ofte 0-0 (nul-nul)-overgange og har samme energi i både absorption og fluorescens.
Udvikling af princippet
I en rapport offentliggjort i 1926 af Transactions of the Faraday Society beskæftigede James Franck sig med mekanismerne i fotoinducerede kemiske reaktioner. Den formodede mekanisme var excitation af et molekyle ved hjælp af en foton efterfulgt af en kollision med et andet molekyle i løbet af den korte periode af excitationen. Spørgsmålet var, om det var muligt for et molekyle at blive opdelt i fotoprodukter i et enkelt trin, nemlig absorptionen af en foton, og uden en kollision. For at et molekyle kan gå i stykker, skal det fra fotonen opnå en vibrationsenergi, der overstiger dissociationsenergien, dvs. den energi, der er nødvendig for at bryde en kemisk binding. Men som man vidste på det tidspunkt, vil molekyler kun absorbere energi svarende til tilladte kvanteovergange, og der findes ikke noget vibrationsniveau over dissociationsenerginiveauet i potentialbrønden. Fotonabsorption med høj energi fører til en overgang til en højere elektronisk tilstand i stedet for dissociation. Ved at undersøge, hvor meget vibrationsenergi et molekyle kan få, når det exciteres til et højere elektronisk niveau, og om denne vibrationsenergi kan være nok til straks at splitte molekylet ad, tegnede han tre diagrammer, der repræsenterer de mulige ændringer i bindingsenergien mellem den laveste elektroniske tilstand og højere elektroniske tilstande.
Diagram I. viser en stor svækkelse af bindingen ved en overgang fra den normale tilstand n til de exciterede tilstande a og a’. Her har vi D > D’ og D’ > D”. Samtidig flytter kernes ligevægtsstilling sig med anspændingen til større værdier af r. Hvis vi går fra ligevægtsstillingen (minimum af potentiel energi) i n-kurven lodret opad til a-kurverne i diagram I. vil partiklerne have en potentiel energi, der er større end D’, og vil flyve fra hinanden. I dette tilfælde har vi en meget stor ændring i svingningsenergien ved excitering med lys… – James Franck 1926
James Franck erkendte, at ændringer i svingningsniveauer kunne være en konsekvens af den øjeblikkelige karakter af excitering til højere elektroniske energiniveauer og en ny ligevægtsposition for det nukleare interaktionspotentiale. Edward Condon udvidede denne indsigt ud over fotoreaktioner i en artikel i Physical Review fra 1926 med titlen “A Theory of Intensity Distribution in Band Systems”. Her formulerer han den semiklassiske formulering på en måde, der minder meget om dens moderne form. Den første fælles henvisning til både Franck og Condon med hensyn til det nye princip findes i samme nummer af Physical Review fra 1926 i en artikel om båndstrukturen i kulilte af Raymond Birge.
Kvantemekanisk formulering
Og tænk på en elektrisk dipolovergang fra den indledende vibrationstilstand (ʋ) af det elektroniske grundniveau (ε), 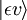 , til en vis vibrationstilstand (ʋ’ ) af en exciteret elektronisk tilstand (ε’),
, til en vis vibrationstilstand (ʋ’ ) af en exciteret elektronisk tilstand (ε’), 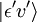 (se bra-ket notation). Den molekylære dipoloperator μ er bestemt af elektronernes ladninger (-e) og placeringer (ri) samt kernes ladninger (+eZj) og placeringer (Rj):
(se bra-ket notation). Den molekylære dipoloperator μ er bestemt af elektronernes ladninger (-e) og placeringer (ri) samt kernes ladninger (+eZj) og placeringer (Rj):
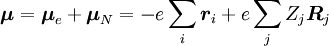
Sandsynlighedsamplituden P for overgangen mellem disse to tilstande er givet ved
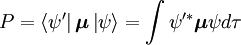
Hvor 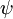 og
og 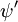 er henholdsvis de samlede bølgefunktioner for den indledende og den endelige tilstand. De samlede bølgefunktioner er produktet af de individuelle vibrations- (afhængig af kernes rumkoordinater) og elektroniske rum- og spinbølgefunktioner:
er henholdsvis de samlede bølgefunktioner for den indledende og den endelige tilstand. De samlede bølgefunktioner er produktet af de individuelle vibrations- (afhængig af kernes rumkoordinater) og elektroniske rum- og spinbølgefunktioner:
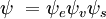
Denne adskillelse af de elektroniske og vibrationsbølgefunktioner er et udtryk for Born-Oppenheimer-approksimationen og er den grundlæggende antagelse i Franck-Condon-princippet. Kombinationen af disse ligninger fører til et udtryk for sandsynlighedsamplituden i form af separate elektroniske rum-, spin- og vibrationsbidrag:
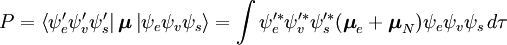
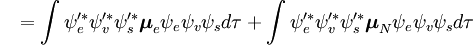
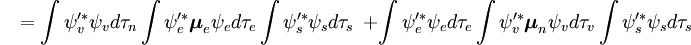
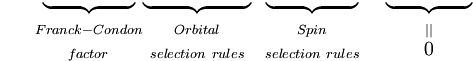
Den spin-uafhængige del af det indledende integral er her tilnærmet som et produkt af to integraler
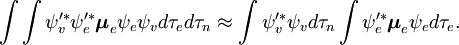
Denne faktorisering ville være nøjagtig, hvis integralet 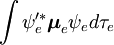 over elektronernes rumkoordinater ikke ville afhænge af kernekoordinaterne. I Born-Oppenheimer-approksimationen afhænger
over elektronernes rumkoordinater ikke ville afhænge af kernekoordinaterne. I Born-Oppenheimer-approksimationen afhænger 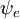 og
og 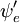 imidlertid (parametrisk) af kernekoordinaterne, således at integralet (en såkaldt overgangsdipolflade) er en funktion af kernekoordinaterne. Da afhængigheden normalt er ret jævn, er det ofte tilladt at negligere den (dvs. at antage, at overgangsdipolfladen er uafhængig af kernekoordinaterne).
imidlertid (parametrisk) af kernekoordinaterne, således at integralet (en såkaldt overgangsdipolflade) er en funktion af kernekoordinaterne. Da afhængigheden normalt er ret jævn, er det ofte tilladt at negligere den (dvs. at antage, at overgangsdipolfladen er uafhængig af kernekoordinaterne).
Det første integral efter plustegnet er lig med nul, fordi de elektroniske bølgefunktioner for forskellige tilstande er ortogonale. Tilbage er produktet af tre integraler. Det første integral er det vibrationsmæssige overlapningsintegral, også kaldet Franck-Condon-faktoren. De resterende to integraler, der bidrager til sandsynlighedsamplituden, bestemmer de elektroniske rum- og spinudvælgelsesregler.
Frank-Condon-princippet er et udsagn om tilladte vibrationsovergange mellem to forskellige elektroniske tilstande, andre kvantemekaniske udvælgelsesregler kan sænke sandsynligheden for en overgang eller helt forbyde den. Rotationelle udvælgelsesregler er blevet negligeret i ovenstående afledning. Rotationsbidrag kan observeres i gasers spektrer, men er stærkt undertrykt i væsker og faste stoffer.
Det bør være klart, at den kvantemekaniske formulering af Franck-Condon-princippet er resultatet af en række tilnærmelser, hovedsageligt antagelsen om den elektriske dipolovergang og Born-Oppenheimer-approksimationen. Svagere magnetiske dipol- og elektriske quadrupol-elektroniske overgange sammen med den ufuldstændige gyldighed af faktoriseringen af den samlede bølgefunktion i nukleare, elektroniske rumlige og spin-bølgefunktioner betyder, at udvælgelsesreglerne, herunder Franck-Condon-faktoren, ikke overholdes strengt. For en given overgang bestemmes værdien af P af alle udvælgelsesreglerne, men spinudvælgelse er den største bidragyder, efterfulgt af elektroniske udvælgelsesregler. Franck-Condon-faktoren modulerer kun svagt intensiteten af overgangene, dvs. den bidrager med en faktor i størrelsesordenen 1 til intensiteten af bånd, hvis størrelsesorden er bestemt af de andre udvælgelsesregler. Nedenstående tabel viser intervallet af extinktionskoefficienter for de mulige kombinationer af tilladte og forbudte spin- og orbitaludvælgelsesregler.
103 til 105
100 til 103
10-5 til 100
Franck-Condon-metaforer i spektroskopi
Det Franck-Condon-princip, i sin kanoniske form, gælder kun for ændringer i et molekyls vibrationsniveauer i forbindelse med en ændring i de elektroniske niveauer ved enten absorption eller emission af en foton. Den fysiske intuition af dette princip er forankret i tanken om, at atomernes kernekoordinater, der udgør molekylet, ikke har tid til at ændre sig i løbet af den meget korte tid, der er involveret i en elektronisk overgang. Denne fysiske intuition kan imidlertid udvides til at omfatte vekselvirkninger mellem lysabsorberende eller lysemitterende molekyler (chromophorer) og deres omgivelser, og det sker faktisk rutinemæssigt. Franck-Condon-metaforen er hensigtsmæssig, fordi molekyler ofte interagerer kraftigt med de omkringliggende molekyler, især i væsker og faste stoffer, og disse interaktioner ændrer kromoforens kernekoordinater på en måde, der er tæt analog med de molekylære vibrationer, som Franck-Condon-princippet tager højde for.
Franck-Condon-princippet for fononer
Den nærmeste Franck-Condon-analogi skyldes interaktionen mellem fononer -kvanter af gittervibrationer- med de elektroniske overgange af kromoforer, der er indlejret som urenheder gitteret. I denne situation kan der ske overgange til højere elektroniske niveauer, når fotonens energi svarer til den rent elektroniske overgangsenergi eller til den rent elektroniske overgangsenergi plus energien fra en eller flere gitterfononer. I lavtemperaturapproksimationen sker emissionen fra nul-fononniveauet i den exciterede tilstand til nul-fonniveauet i grundtilstanden eller til højere fononniveauer i grundtilstanden. Ligesom i Franck-Condon-princippet bestemmes sandsynligheden for overgange, der involverer fononer, af overlapningen af fononbølgefunktionerne ved det indledende og det endelige energiniveau. For Franck-Condon-princippet anvendt på fononovergange er etiketten på den vandrette akse i figur 1 i figur 6 erstattet med konfigurations-koordinaten for en normal tilstand. Den potentielle energi for gittertilstanden qi i figur 6 er repræsenteret som den potentielle energi for en harmonisk oscillator, og afstanden mellem foneniveauerne (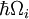 ) er bestemt af gitterparametre. Da energien af enkelte fononer generelt er ret lille, kan nul- eller få-fonononovergange kun observeres ved temperaturer under ca. 40 kelvin.
) er bestemt af gitterparametre. Da energien af enkelte fononer generelt er ret lille, kan nul- eller få-fonononovergange kun observeres ved temperaturer under ca. 40 kelvin.
Se nul-fononlinje og fonon-sidebånd for yderligere oplysninger og referencer.
Franck-Condon-princippet i solvens
Franck-Condon-overvejelser kan også anvendes på de elektroniske overgange hos chromophorer, der er opløst i væsker. I denne anvendelse af Franck-Condon-metaforen bidrager kromoforernes vibrationsniveauer samt kromoforernes vekselvirkninger med fononer i væsken fortsat til strukturen af absorptions- og emissionsspektre, men disse virkninger betragtes separat og uafhængigt.
Tænk på kromoforer omgivet af opløsningsmiddelmolekyler. Disse omgivende molekyler kan interagere med kromohorerne, især hvis opløsningsmiddelmolekylerne er polære. Denne forbindelse mellem opløsningsmiddel og opløst stof kaldes solvering og er en stabiliserende vekselvirkning, dvs. at opløsningsmiddelmolekylerne kan bevæge sig og rotere, indtil vekselvirkningsenergien er minimeret. Selve vekselvirkningen omfatter elektrostatiske kræfter og van der Waals-kræfter og kan også omfatte hydrogenbindinger. Franck-Condon-principperne kan anvendes, når vekselvirkningerne mellem chromoforen og de omgivende opløsningsmiddelmolekyler er forskellige i den grundtilstand og i den exciterede elektroniske tilstand. Denne ændring i vekselvirkningen kan f.eks. skyldes forskellige dipolmomenter i disse to tilstande. Hvis kromosoffen starter i grundtilstanden og er tæt på ligevægt med de omgivende opløsningsmiddelmolekyler og derefter absorberer en foton, der bringer den til den exciterede tilstand, vil dens vekselvirkning med opløsningsmidlet være langt fra ligevægt i den exciterede tilstand. Denne effekt svarer til det oprindelige Franck-Condon-princip: den elektroniske overgang er meget hurtig i forhold til kernes bevægelse – omgruppering af opløsningsmiddelmolekyler i tilfælde af solvens -. Vi taler nu om en vertikal overgang, men nu er den horisontale koordinat opløsningsmiddel-solut-interaktionsrummet. Denne koordinatakse betegnes ofte som “Solvationskoordinat” og repræsenterer, lidt abstrakt, alle de relevante bevægelsesdimensioner for alle de vekselvirkende opløsningsmolekyler.
I det oprindelige Franck-Condon-princip begynder de molekyler, som ender i højere vibrationstilstande, efter den elektroniske overgang straks at slappe af til den laveste vibrationstilstand. I tilfælde af solvering vil opløsningsmolekylerne straks forsøge at omarrangere sig selv for at minimere vekselvirkningsenergien. Hastigheden af opløsningsmiddelrelaksation afhænger af opløsningsmidlets viskositet. Hvis man antager, at opløsningsmidlets afslapningstid er kort sammenlignet med levetiden for den elektroniske exciterede tilstand, vil emissionen ske fra den exciterede elektroniske tilstands laveste energitilstand i opløsningsmidlet. For småmolekylære opløsningsmidler som f.eks. vand eller methanol ved omgivelsestemperatur er opløsningsmiddelrelaksationstiden i størrelsesordenen nogle tiendedele picosekunder, mens levetiden for chromoforexciterede tilstande varierer fra et par picosekunder til et par nanosekunder. Umiddelbart efter overgangen til den elektroniske grundtilstand skal opløsningsmiddelmolekylerne også omarrangere sig selv for at tilpasse sig kromoforens nye elektroniske konfiguration. Figur 7 illustrerer Franck-Condon-princippet anvendt på solvering. Når opløsningen belyses med lys, der svarer til den elektroniske overgangsenergi, vil nogle af kromoforerne bevæge sig til den exciterede tilstand. Inden for denne gruppe af chromophorer vil der være en statistisk fordeling af vekselvirkningsenergierne mellem opløsningsmiddel og chromophorer, som i figuren er repræsenteret ved en Gauss-fordelingsfunktion. Vekselvirkningen mellem opløsningsmiddel og chromophor er tegnet som et parabolisk potentiale i begge elektroniske tilstande. Da den elektroniske overgang stort set er øjeblikkelig på tidsskalaen for opløsningsmidlets bevægelse (lodret pil), er samlingen af chromophorer i exciteret tilstand umiddelbart langt fra ligevægt. Opløsningsmiddelmolekylernes omlægning i henhold til den nye potentialeenergikurve er repræsenteret ved de buede pile i figur 7. Bemærk, at mens de elektroniske overgange er kvantiserede, behandles vekselvirkningsenergien mellem kromoforen og opløsningsmidlet som et klassisk kontinuum på grund af det store antal molekyler, der er involveret. Selv om emissionen er afbildet som værende foregået fra minimum af interaktionspotentialet mellem kromoforen og opløsningsmidlet i den exciterede tilstand, kan der ske en betydelig emission, før der er opnået ligevægt, hvis opløsningsmidlets viskositet er høj, eller hvis levetiden for den exciterede tilstand er kort. Energiforskellen mellem absorberede og emitterede fotoner, der er vist i figur 7, er solvationsbidraget til Stokesforskydningen.
Journal links kan kræve abonnement.
- Franck, J. (1926). “Elementære processer i fotokemiske reaktioner”. Transactions of the Faraday Society 21: 536-542. Link
- Condon, E. (1926). “A theory of intensity distribution in band systems (Meeting abstract)”. Physical Review 27: 640.
- Condon, E. (1926). “A theory of intensity distribution in band systems”. Physical Review 28: 1182-1201. Link
- Condon, E. (1928). “Nuclear motions associated with electron transitions in diatomic molecules”. Physical Review 32: 858-872. Link
- Birge, R. T. (1926). “The band spectra of carbon monoxide”. Physical Review 28: 1157-1181. Link
- Noyes, W. A. (1933). “The correlation of spectroscopy and photochemistry”. Reviews of Modern Physics 5: 280-287. Link
- Coolidge, A. S, James, H. M. og Present, R. D. (1936). “A study of the Franck-Condon Principle”. Journal of Chemical Physics 4: 193-211. Link
- Herzberg, Gerhard (1971). Spektrer og strukturer af simple frie radikaler. New York: Dover. ISBN 0-486-65821-X.
- Harris, Daniel C.; Michael D. Bertolucci (1978). Symmetri og spektroskopi. New York: Dover. ISBN 0-486-66144-X.
- Bernath, Peter F. (1995). Spectra of Atoms and Molecules (Topics in Physical Chemistry). Oxford: Oxford University Press. ISBN 0-19-507598-6.
- Atkins, P. W.; R. S. Frieman (1999). Molekylær kvantemekanik. Oxford: Oxford University Press. ISBN 0-19-855947-X.
Se også
- Born-Oppenheimer-approksimation
- Molekylær elektronisk overgang
- Ultraviolet-synlig spektroskopi
- Kvanteharmonisk oscillator
- Morsepotentiale
- Vibronisk kobling
- Nul-fononlinje og fonon-sidebånd
- Sudden-approximation
Kategorier: Kvantekemi | Spektroskopi | Molekylærfysik