Zasada Francka-Condona jest zasadą w spektroskopii i chemii kwantowej, która wyjaśnia intensywność przejść wibronowych. Przejścia wibronowe to jednoczesne zmiany poziomów energetycznych elektronowego i wibracyjnego cząsteczki w wyniku absorpcji lub emisji fotonu o odpowiedniej energii. Zasada ta mówi, że podczas przejścia elektronowego, zmiana z jednego wibracyjnego poziomu energetycznego na inny będzie bardziej prawdopodobna, jeśli dwie wibracyjne funkcje falowe pokrywają się w większym stopniu.
Dodatkowa zalecana wiedza
Zawartość
- 1 Przegląd
- 2 Rozwój zasady
- 3 Sformułowanie kwantowo-mechaniczne
- 4 Metafory Francka-Condona w spektroskopii
- 4.1 Zasada Francka-Condona dla fononów
- 4.2 Zasada Francka-Condona w solwatacji
- 5 Referencje
- 6 Zobacz także
Przegląd
Zasada Francka-Condona ma dobrze ugruntowaną semiklasyczną interpretację opartą na oryginalnym wkładzie Jamesa Francka. Przejścia elektroniczne są zasadniczo natychmiastowe w porównaniu ze skalą czasową ruchów jądrowych, dlatego jeśli cząsteczka ma przejść na nowy poziom wibracyjny podczas przejścia elektronowego, ten nowy poziom wibracyjny musi być natychmiast zgodny z pozycjami jądrowymi i momentami pędu poziomu wibracyjnego cząsteczki w wyjściowym stanie elektronowym. W semiklasycznym obrazie drgań (oscylacji) prostego oscylatora harmonicznego konieczne warunki mogą wystąpić w punktach zwrotnych, gdzie pęd jest równy zeru.
Klasycznie zasada Francka-Condona jest przybliżeniem, że przejście elektronowe zachodzi najprawdopodobniej bez zmian położeń jąder w jednostce molekularnej i jej otoczeniu. Powstały w ten sposób stan nazywamy stanem Francka-Condona, a dane przejście przejściem pionowym. Kwantowo-mechaniczne sformułowanie tej zasady mówi, że intensywność przejścia wibronowego jest proporcjonalna do kwadratu całki nakładającej się pomiędzy wibracyjnymi funkcjami falowymi dwóch stanów, które biorą udział w przejściu. – IUPAC Compendium of Chemical Terminology, 2nd Edition (1997)
W obrazie kwantowo-mechanicznym, poziomy wibracyjne i wibracyjne funkcje falowe są poziomami kwantowych oscylatorów harmonicznych lub bardziej złożonych przybliżeń energii potencjalnej cząsteczek, takich jak potencjał Morse’a. Rysunek 1 ilustruje zasadę Francka-Condona dla przejść wibronowych w cząsteczce z funkcjami energii potencjalnej podobnymi do funkcji Morse’a zarówno w podstawowym jak i wzbudzonym stanie elektronowym. W niskotemperaturowym przybliżeniu, cząsteczka zaczyna w poziomie wibracyjnym v = 0 podstawowego stanu elektronowego i po zaabsorbowaniu fotonu o odpowiedniej energii, dokonuje przejścia do wzbudzonego stanu elektronowego. Konfiguracja elektronowa nowego stanu może powodować przesunięcie położenia równowagowego jąder tworzących cząsteczkę. Na rysunku to przesunięcie współrzędnych atomowych pomiędzy stanem podstawowym a pierwszym stanem wzbudzonym oznaczone jest jako q 10. W najprostszym przypadku cząsteczki dwuatomowej oś współrzędnych jądrowych odnosi się do separacji międzyjądrowej. Przejście wibronowe jest zaznaczone pionową strzałką ze względu na założenie stałych współrzędnych jądrowych podczas przejścia. Prawdopodobieństwo, że cząsteczka może znaleźć się na danym poziomie wibracyjnym jest proporcjonalne do kwadratu (pionowego) nakładania się wibracyjnych funkcji falowych stanu początkowego i końcowego (patrz poniżej sekcja Sformułowanie kwantowo-mechaniczne). W elektronowym stanie wzbudzonym cząsteczki szybko relaksują do najniższego poziomu wibracyjnego (reguła Kashy), a stamtąd mogą rozpadać się do najniższego stanu elektronowego poprzez emisję fotonów. Zasada Francka-Condona jest stosowana zarówno do absorpcji jak i do fluorescencji.
Zastosowanie zasady Francka-Condona zarówno w absorpcji jak i we fluorescencji, wraz z regułą Kashy prowadzi do przybliżonej symetrii lustrzanej pokazanej na rysunku 2. Struktura wibracyjna molekuł w zimnym, rzadkim gazie jest najlepiej widoczna ze względu na brak niejednorodnego poszerzenia poszczególnych przejść. Przejścia wibracyjne narysowane są na rysunku 2 jako wąskie, równomiernie rozłożone kształty linii Lorentziana. Równe odstępy pomiędzy poziomami wibracyjnymi występują tylko w przypadku parabolicznego potencjału prostych oscylatorów harmonicznych, w bardziej realistycznych potencjałach, takich jak te przedstawione na rysunku 1, odstępy energetyczne maleją wraz ze wzrostem energii wibracji. Przejścia elektronowe do i z najniższych stanów wibracyjnych są często określane jako przejścia 0-0 (zero zero zero) i mają taką samą energię zarówno w absorpcji jak i fluorescencji.
Rozwój zasady
W raporcie opublikowanym w 1926 roku w Transactions of the Faraday Society, James Franck zajmował się mechanizmami reakcji chemicznych indukowanych fotonami. Zakładany mechanizm polegał na wzbudzeniu cząsteczki przez foton, a następnie zderzeniu z inną cząsteczką podczas krótkiego okresu wzbudzenia. Pytanie brzmiało, czy jest możliwe, aby cząsteczka rozpadła się na fotoprodukty w jednym kroku – absorpcji fotonu i bez zderzenia. Aby cząsteczka mogła się rozpadać, musi otrzymać od fotonu energię drgań przekraczającą energię dysocjacji, czyli energię do rozerwania wiązania chemicznego. Jednak, jak wówczas wiedziano, cząsteczki zaabsorbują tylko energię odpowiadającą dozwolonym przejściom kwantowym i nie istnieją poziomy drgań powyżej poziomu energii dysocjacji studni potencjału. Absorpcja fotonu o wysokiej energii prowadzi do przejścia do wyższego stanu elektronowego zamiast do dysocjacji. Badając, jak dużą energię wibracyjną może uzyskać cząsteczka, gdy jest wzbudzona do wyższego poziomu elektronowego i czy ta energia wibracyjna może być wystarczająca do natychmiastowego rozpadu cząsteczki, narysował trzy diagramy przedstawiające możliwe zmiany energii wiązania pomiędzy najniższym stanem elektronowym a wyższymi stanami elektronowymi.
Diagram I. pokazuje duże osłabienie wiązania przy przejściu ze stanu normalnego n do stanów wzbudzonych a i a’. Mamy tu D > D’ i D’ > D”. Jednocześnie położenie równowagowe jąder przesuwa się wraz ze wzbudzeniem do większych wartości r. Jeżeli przejdziemy od położenia równowagowego (minimum energii potencjalnej) krzywej n pionowo w górę do krzywych a na schemacie I., to cząsteczki będą miały energię potencjalną większą niż D’ i rozlecą się. W tym przypadku mamy bardzo dużą zmianę w energii oscylacji przy wzbudzeniu światłem… – James Franck 1926
James Franck uznał, że zmiany w poziomach wibracyjnych mogą być konsekwencją natychmiastowej natury wzbudzenia do wyższych elektronicznych poziomów energetycznych i nowego położenia równowagi dla potencjału oddziaływania jądrowego. Edward Condon rozszerzył to spostrzeżenie poza fotoreakcje w artykule z 1926 r. w Physical Review zatytułowanym „A Theory of Intensity Distribution in Band Systems”. Sformułował on tam semiklasyczne sformułowanie w sposób dość podobny do jego współczesnej postaci. Pierwsze wspólne odniesienie do Francka i Condona w odniesieniu do nowej zasady pojawia się w tym samym numerze Physical Review z 1926 roku w artykule Raymonda Birge’a o strukturze pasmowej tlenku węgla.
Sformułowanie kwantowo-mechaniczne
Rozważmy elektryczne przejście dipolowe z początkowego stanu wibracyjnego (ʋ) podstawowego poziomu elektronowego (ε), |epsilon v’‖3769>, do pewnego stanu wibracyjnego (ʋ’ ) wzbudzonego stanu elektronowego (ε’), |epsilon’ v’‖9703> (patrz notacja bra-ket). Molekularny operator dipolowy μ jest określony przez ładunki (-e) i położenia (ri) elektronów oraz ładunki (+eZj) i położenia (Rj) jąder atomowych:
\Boldsymbol{\mu} = \boldsymbol{\mu} _e + \boldsymbol{\mu} _N = – e\u>suma\u_i {\boldsymbol{r}_i } \gdzie i są, odpowiednio, ogólnymi funkcjami falowymi stanu początkowego i końcowego. Połączenie tych równań prowadzi do wyrażenia amplitudy prawdopodobieństwa w kategoriach oddzielnych wkładów przestrzeni elektronowej, spinowej i wibracyjnej: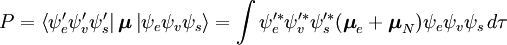
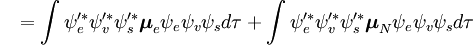
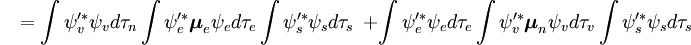
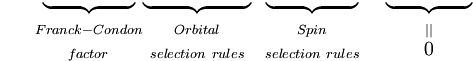
Niezależna od spinu część całki początkowej jest tu aproksymowana jako iloczyn dwóch całek
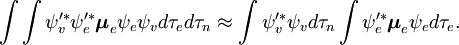
Faktoryzacja ta byłaby dokładna, gdyby całka 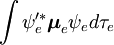 po współrzędnych przestrzennych elektronów nie zależałaby od współrzędnych jądrowych. Jednak w przybliżeniu Borna-Oppenheimera
po współrzędnych przestrzennych elektronów nie zależałaby od współrzędnych jądrowych. Jednak w przybliżeniu Borna-Oppenheimera 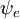 i
i 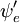 zależ± (parametrycznie) od współrzędnych j±drowych, tak że całka (tzw. powierzchnia dipolowa przej¶cia) jest funkcj± współrzędnych j±drowych. Ponieważ zależność ta jest zwykle dość gładka, często dopuszcza się jej zaniedbanie (tzn. założenie, że powierzchnia dipola przejściowego jest niezależna od współrzędnych jądrowych).
zależ± (parametrycznie) od współrzędnych j±drowych, tak że całka (tzw. powierzchnia dipolowa przej¶cia) jest funkcj± współrzędnych j±drowych. Ponieważ zależność ta jest zwykle dość gładka, często dopuszcza się jej zaniedbanie (tzn. założenie, że powierzchnia dipola przejściowego jest niezależna od współrzędnych jądrowych).
Pierwsza całka po znaku plus jest równa zero, ponieważ funkcje falowe elektronów różnych stanów są ortogonalne. Pozostała część jest iloczynem trzech całek. Pierwsza całka jest całką nakładania drgań, zwaną też współczynnikiem Francka-Condona. Pozostałe dwie całki składające się na amplitudę prawdopodobieństwa określają elektroniczne przestrzenne i spinowe reguły selekcji.
Zasada Francka-Condona jest stwierdzeniem o dozwolonych przejściach wibracyjnych pomiędzy dwoma różnymi stanami elektronowymi, inne kwantowo-mechaniczne reguły selekcji mogą obniżać prawdopodobieństwo przejścia lub całkowicie go zabraniać. Rotacyjne reguły selekcji zostały pominięte w powyższym wyprowadzeniu. Wkład rotacyjny może być obserwowany w widmach gazów, ale jest silnie tłumiony w cieczach i ciałach stałych.
Powinno być jasne, że kwantowo-mechaniczne sformułowanie zasady Francka-Condona jest wynikiem serii przybliżeń, głównie założenia o elektrycznym przejściu dipolowym i przybliżenia Borna-Oppenheimera. Słabsze przejścia elektroniczne dipol magnetyczny i kwadrupol elektryczny oraz niepełna słuszność faktoryzacji całkowitej funkcji falowej na funkcje falowe jądrowe, elektroniczne przestrzenne i spinowe powodują, że reguły selekcji, w tym współczynnik Francka-Condona, nie są ściśle przestrzegane. Dla danego przejścia wartość P jest zdeterminowana przez wszystkie reguły selekcji, przy czym największy udział ma selekcja spinowa, a następnie elektronowa. Czynnik Francka-Condona tylko słabo moduluje intensywność przejść, tzn. wnosi czynnik rzędu 1 do intensywności pasm, których rząd wielkości jest określony przez pozostałe reguły selekcji. W tabeli poniżej podano zakres współczynników ekstynkcji dla możliwych kombinacji dozwolonych i zabronionych reguł selekcji spinowej i orbitalnej.
103 do 105
100 do 103
10-5 do 100
Metafory Francka-Condona w spektroskopii
Zasada Francka-Condona, w swojej kanonicznej postaci dotyczy tylko zmian poziomów wibracyjnych cząsteczki w trakcie zmiany poziomów elektronowych poprzez absorpcję lub emisję fotonu. Fizyczna intuicja tej zasady opiera się na założeniu, że współrzędne jądrowe atomów tworzących cząsteczkę nie mają czasu na zmianę podczas bardzo krótkiego czasu związanego z przejściem elektronowym. Jednakże, ta fizyczna intuicja może być, i rzeczywiście jest, rutynowo rozszerzona na interakcje pomiędzy absorbującymi lub emitującymi światło cząsteczkami (chromoforami) a ich otoczeniem. Metafory Francka-Condona są odpowiednie, ponieważ molekuły często silnie oddziałują z otaczającymi je molekułami, szczególnie w cieczach i ciałach stałych, a te oddziaływania modyfikują współrzędne jądrowe chromoforu w sposób, który jest ściśle analogiczny do drgań molekularnych rozważanych przez zasadę Francka-Condona.
Zasada Francka-Condona dla fononów
Najbliższa analogia Francka-Condona wynika z oddziaływania fononów – kwantów drgań sieci – z przejściami elektronowymi chromoforów osadzonych jako zanieczyszczenia sieci. W tej sytuacji przejście na wyższe poziomy elektronowe może nastąpić, gdy energia fotonu odpowiada czysto elektronowej energii przejścia lub czysto elektronowej energii przejścia powiększonej o energię jednego lub kilku fononów sieci. W niskotemperaturowym przybliżeniu emisja następuje z zerowego poziomu fonowego stanu wzbudzonego do zerowego poziomu fonowego stanu podstawowego lub do wyższych poziomów fonowych stanu podstawowego. Podobnie jak w zasadzie Francka-Condona, prawdopodobieństwo przejść z udziałem fononów jest określone przez nakładanie się fonowych funkcji falowych na początkowym i końcowym poziomie energetycznym. Dla zasady Francka-Condona zastosowanej do przejść fonowych, etykieta osi poziomej na Rysunku 1 jest zastąpiona na Rysunku 6 współrzędną konfiguracyjną dla trybu normalnego. Energia potencjalna qi trybu sieciowego na rysunku 6 jest przedstawiona jako energia oscylatora harmonicznego, a odstępy pomiędzy poziomami fononów (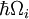 ) są określone przez parametry sieci. Ponieważ energia pojedynczych fononów jest zwykle dość mała, przejścia zero- lub kilka fononów można zaobserwować tylko w temperaturach poniżej około 40 kelwinów.
) są określone przez parametry sieci. Ponieważ energia pojedynczych fononów jest zwykle dość mała, przejścia zero- lub kilka fononów można zaobserwować tylko w temperaturach poniżej około 40 kelwinów.
Zobacz Zero-phononon line i fon sideband dla dalszych szczegółów i odniesienia.
Zasada Francka-Condona w solwatacji
Rozważania Francka-Condona mogą być również zastosowane do przejść elektronowych chromoforów rozpuszczonych w cieczach. W tym zastosowaniu metafory Francka-Condona, poziomy wibracyjne chromoforów, jak również oddziaływania chromoforów z fononami w cieczy, nadal przyczyniają się do struktury widm absorpcji i emisji, ale efekty te są rozważane oddzielnie i niezależnie.
Rozważmy chromofory otoczone przez cząsteczki rozpuszczalnika. Te otaczające cząsteczki mogą oddziaływać z chromoforami, szczególnie jeśli cząsteczki rozpuszczalnika są polarne. Ta asocjacja pomiędzy rozpuszczalnikiem i solutem jest określana jako solwatacja i jest oddziaływaniem stabilizującym, to znaczy, że cząsteczki rozpuszczalnika mogą się poruszać i obracać, aż energia oddziaływania zostanie zminimalizowana. Samo oddziaływanie obejmuje siły elektrostatyczne i siły van der Waalsa, a także może obejmować wiązania wodorowe. Zasady Francka-Condona mogą być stosowane, gdy oddziaływania pomiędzy chromoforem a otaczającymi go cząsteczkami rozpuszczalnika są różne w podstawowym i wzbudzonym stanie elektronowym. Ta zmiana oddziaływania może wynikać np. z różnych momentów dipolowych w tych dwóch stanach. Jeśli chromofor zaczyna w swoim stanie podstawowym i jest bliski równowagi z otaczającymi go cząsteczkami rozpuszczalnika, a następnie absorbuje foton, który przenosi go do stanu wzbudzonego, jego oddziaływanie z rozpuszczalnikiem będzie dalekie od równowagi w stanie wzbudzonym. Efekt ten jest analogiczny do pierwotnej zasady Francka-Condona: przejście elektronowe jest bardzo szybkie w porównaniu z ruchem jąder – rearanżacją cząsteczek rozpuszczalnika w przypadku solwatacji. Mówimy teraz o przejściu pionowym, ale teraz współrzędną poziomą jest przestrzeń oddziaływań rozpuszczalnik-rozpuszczalnik. Ta oś współrzędnych jest często oznaczana jako „współrzędna solwatacji” i reprezentuje, nieco abstrakcyjnie, wszystkie istotne wymiary ruchu wszystkich oddziałujących cząsteczek rozpuszczalnika.
W oryginalnej zasadzie Francka-Condona, po przejściu elektronowym, cząsteczki, które kończą w wyższych stanach wibracyjnych natychmiast zaczynają relaksować do najniższego stanu wibracyjnego. W przypadku solwatacji, cząsteczki rozpuszczalnika będą natychmiast starały się zmienić swój układ w celu zminimalizowania energii oddziaływania. Szybkość relaksacji rozpuszczalnika zależy od jego lepkości. Zakładając, że czas relaksacji rozpuszczalnika jest krótki w porównaniu z czasem życia elektronowego stanu wzbudzonego, emisja będzie następowała z najniższego stanu energetycznego wzbudzonego stanu elektronowego w rozpuszczalniku. W przypadku rozpuszczalników małocząsteczkowych, takich jak woda czy metanol w temperaturze otoczenia, czas relaksacji rozpuszczalnika jest rzędu kilkudziesięciu pikosekund, podczas gdy czasy życia stanu wzbudzonego chromoforu wynoszą od kilku pikosekund do kilku nanosekund. Natychmiast po przejściu do podstawowego stanu elektronowego, cząsteczki rozpuszczalnika muszą również zmienić swój układ, aby dostosować się do nowej konfiguracji elektronowej chromoforu. Rysunek 7 ilustruje zasadę Francka-Condona zastosowaną do solwatacji. Gdy roztwór zostanie oświetlony światłem o energii odpowiadającej energii przejścia elektronowego, niektóre z chromoforów przejdą do stanu wzbudzonego. W tej grupie chromoforów będzie istniał statystyczny rozkład energii oddziaływania rozpuszczalnik-chromofor, reprezentowany na rysunku przez gaussowską funkcję rozkładu. Oddziaływanie rozpuszczalnik-chromofor jest narysowane jako potencjał paraboliczny w obu stanach elektronicznych. Ponieważ przejście elektronowe jest w zasadzie natychmiastowe w skali czasowej ruchu rozpuszczalnika (pionowa strzałka), zbiór chromoforów w stanie wzbudzonym jest natychmiast daleki od równowagi. Zmiana ułożenia cząsteczek rozpuszczalnika zgodnie z nową krzywą energii potencjalnej jest przedstawiona za pomocą zakrzywionych strzałek na Rysunku 7. Zauważmy, że podczas gdy przejścia elektroniczne są skwantowane, energia oddziaływania chromofor-rozpuszczalnik jest traktowana jako klasyczne kontinuum ze względu na dużą liczbę zaangażowanych cząsteczek. Chociaż emisja jest przedstawiona jako zachodząca od minimum potencjału oddziaływania chromofor-rozpuszczalnik w stanie wzbudzonym, znaczna emisja może mieć miejsce przed osiągnięciem równowagi, gdy lepkość rozpuszczalnika jest wysoka lub czas życia stanu wzbudzonego jest krótki. Różnica energii pomiędzy zaabsorbowanymi i wyemitowanymi fotonami przedstawiona na Rysunku 7 jest wkładem solwatacji do przesunięcia Stokesa.
Łącza do czasopism mogą wymagać subskrypcji.
- Franck, J. (1926). „Elementarne procesy reakcji fotochemicznych”. Transactions of the Faraday Society 21: 536-542. Link
- Condon, E. (1926). „A theory of intensity distribution in band systems (Meeting abstract)”. Physical Review 27: 640.
- Condon, E. (1926). „A theory of intensity distribution in band systems” (Teoria rozkładu natężenia w układach pasmowych). Physical Review 28: 1182-1201. Link
- Condon, E. (1928). „Nuclear motions associated with electron transitions in diatomic molecules”. Physical Review 32: 858-872. Link
- Birge, R. T. (1926). „The band spectra of carbon monoxide”. Physical Review 28: 1157-1181. Link
- Noyes, W. A. (1933). „Korelacja spektroskopii i fotochemii”. Reviews of Modern Physics 5: 280-287. Link
- Coolidge, A. S, James, H. M. i Present, R. D. (1936). „A study of the Franck-Condon Principle”. Journal of Chemical Physics 4: 193-211. Link
- Herzberg, Gerhard (1971). The spectra and structures of simple free radicals. New York: Dover. ISBN 0-486-65821-X.
- Harris, Daniel C.; Michael D. Bertolucci (1978). Symetria i spektroskopia. New York: Dover. ISBN 0-486-66144-X.
- Bernath, Peter F. (1995). Spectra of Atoms and Molecules (Topics in Physical Chemistry). Oxford: Oxford University Press. ISBN 0-19-507598-6.
- Atkins, P. W.; R. S. Frieman (1999). Molecular Quantum Mechanics. Oxford: Oxford University Press. ISBN 0-19-855947-X.
Zobacz też
- Przybliżenie Borna-Oppenheimera
- Molekularne przejścia elektronowe
- Spektroskopia ultrafioletu-.widzialna spektroskopia
- Kwantowy oscylator harmoniczny
- Potencjał Morse’a
- Sprzężenie wibronowe
- Linia zero-fononowa i boczne pasmo fonowe
- Przybliżenie nagłe
Kategorie: Chemia kwantowa | Spektroskopia | Fizyka molekularna
.